Listen to this story:
0:00
/

A meeting in Texas reckons with the future of treatment, following two setbacks in 2020.
O
n a rainy day in November 2020, in a cramped office in a WeWork coworking space in midtown Manhattan, Jeremy Levin, chief executive officer of Ovid Therapeutics, hung up the phone. His company’s chief medical officer had just told him to gather the team; results from the company’s pivotal trial were ready, and they needed to talk through the data. After six years of intense research and an enormous amount of money — including $10 million of his family’s own — Levin was about to learn how his company’s treatment for Angelman syndrome had performed against placebo.Levin collected his colleagues in a tiny, glass-walled conference room. On everyone’s mind was the Angelman community — those with the syndrome frequently need a caregiver throughout their lives; they can have seizures, limited mobility and minimal speech. There was nothing approved for the syndrome, but early data from Ovid’s trials of gaboxadol, a drug originally developed to treat insomnia, hinted that it could improve sleep in those with the syndrome. Even that small change could drastically improve someone’s quality of life.
These thoughts were set against the backdrop of a recent disappointment in the field. Just one month prior, clinical trial participants receiving the highest dose of GTX-102, a new therapeutic for Angelman syndrome from Florida-based startup GeneTx in partnership with pharmaceutical company Ultragenyx, had reported a temporary side effect of weak limbs and an inability to walk. The U.S. Food and Drug Administration (FDA) had put the trial on hold.
Ovid’s chief medical officer called into the conference room. He got straight to the point: There was no statistically significant benefit for children who received gaboxadol versus a placebo. The treatment had failed.
The room fell silent. Levin thought about how the company would need to restructure its programs and how its stock would be battered. But mostly he thought about the families and the overwhelming disappointment they would feel.
Levin began to cry. His colleagues, many of whom had also devoted years of their lives to this project, cried too.
New angle: After trials for its drug gaboxadol failed, Ovid Therapeutics began exploring an alternative approach to Angelman syndrome treatments.
T
he vibe in the lobby of the Kalahari Resort in Round Rock, Texas — a quickly growing city just minutes from Austin — evoked vacation. A couple carrying ‘to-go’ cocktails strolled by in flip-flops, and giggling children played on luggage carts. Rebecca Burdine was not there on vacation, however. During this first week in August 2022, the resort was hosting the annual Angelman Syndrome Foundation meeting, bringing together researchers, drug developers and families of children with Angelman syndrome.Burdine bridges all three types of conference-goers. She is a molecular biologist and was on a steering committee for Ovid’s gaboxadol trial, and she is mother to 16-year-old Sophie, who has Angelman syndrome. Burdine was at the conference in part to present a keynote address and moderate a panel.
It was the first in-person meeting for the community since 2019, and watching the children with Angelman syndrome walking in the lobby and gesturing to their parents, Burdine was reminded of Sophie, who is never far from her mind anyway. Sophie uses a wheelchair and cannot speak, but the field had progressed in the year and a half since the failure of gaboxadol, and Burdine, like the rest of the attendees, was cautiously optimistic about the future of treatment.

On Monday, the first day of the conference, in a wing of the resort far from the swimming pool and arcade, researchers bubbled with that optimism as they discussed the field’s latest findings. Much of the excitement revolved around work on antisense oligonucleotides (ASOs), short strands of genetic code that, when injected into the spinal cord, can alter protein expression in the brain.
People with Angelman syndrome have mutations in or a deletion of the maternal copy of the UBE3A gene. Since the paternal version of the gene is typically silent in the brain, the result is a complete lack of UBE3A protein. ASOs for Angelman syndrome, such as Ultragenyx’s drug GTX-102, unsilence paternal UBE3A with the goal of rescuing protein levels. This approach represents a significant advancement in therapeutics for the condition — and a reason for the field’s buoyancy: Gaboxadol was identified as a potential drug for Angelman because of its ability to balance brain activity, but an ASO could target the syndrome’s root cause.
GTX-102 was the first ASO to reach clinical trials. After nearly a year spent investigating the paralysis, the FDA lifted the clinical hold, agreeing the issue was temporary and likely caused by inflammation at the injection site. The company had updated its protocol to limit inflammation and use a lower starting dosage, and it was at the conference to share preliminary data from the restarted phase 1 and 2 trial. Children who received injections of GTX-102, Ultragenyx said, showed improvements on multiple measures, including assessments of motor function, communication and sleep.
Pharmaceutical company Roche is also working on an ASO and presented new research, highlighting how its approach produces long-lasting UBE3A protein in monkeys’ brains — results that seem to bode well for their ongoing phase 1 trial.
Burdine served as moderator for the sessions. Some of the results were exciting, but she could not help but feel a terrible sense of deja vu. As someone close to the gaboxadol trial, Burdine had seen early data that looked promising, and yet that trial had ended in disappointment.
For the gaboxadol trial, Ovid established a new Clinical Global Impression-Improvement Scale (CGI-I) specifically for Angelman syndrome, and Burdine helped present it to the FDA. The assessment involves trained clinicians comparing a child’s initial ability — such as how well they walk or communicate — with their ability after treatment in the clinical trial. It helps control for the wide range of baseline skills among people with Angelman syndrome. Roche developed its own CGI-I for its trials, which biotech company Ionis will also use for the ASO it has in the works. Ultragenyx developed a CGI-I, too, and will compare results using that one and the Roche version.
One problem, Burdine knew, is that even updated forms of assessment might not capture all of the ways a treatment could benefit a child. Even worse, treatments for many neurodevelopmental conditions, including Angelman syndrome, remain frustratingly susceptible to the placebo effect in clinical trials. Simply enrolling a child in a trial — giving them more medical and parental attention — can cause improvement in some skills. Burdine knew that in the gaboxadol trial, a child who had never walked before took their first steps — but it turned out that they were receiving placebo. That experience told Burdine that, until placebo-controlled trials were run, it would be impossible to know how well a treatment worked.
Burdine thanked the researchers for their presentations and then, her voice wavering, implored the attendees to keep discussing the science — and to not settle for anything other than real, strong effects.
“These kids deserve the best,” she said, and sent everyone off to the dinner reception.
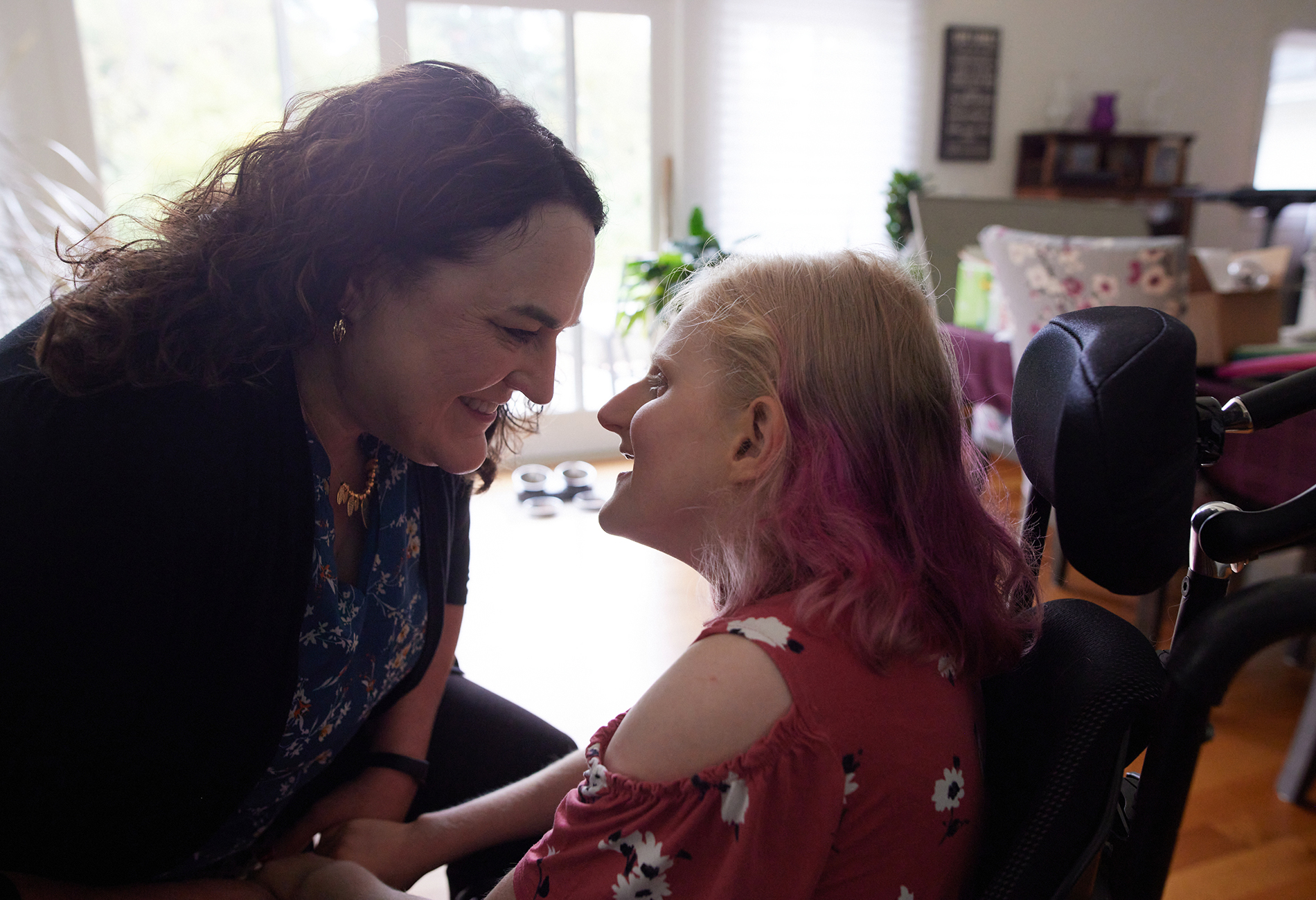
Forging ahead: Treatments for people with Angelman syndrome, such as Rebecca Burdine’s 16-year-old daughter Sophie, have progressed in the 18 months following gaboxadol’s failure.
O
ne way to sidestep the placebo effect is to rely on objective biomarkers. People with Angelman syndrome seem to have atypical electroencephalography (EEG) signals, previous studies show. A recent study found that, in people with the syndrome, the strength of those signals correlates with cognitive skills — suggesting that a drug that normalizes the signals could lead to improvement in other aspects of brain function.At the conference, a researcher presented a new study showing that an ASO that corrects the EEG signals also improves sleep in a mouse model of Angelman syndrome, but whether the same will hold true in people is unknown.
Early data from the paused GTX-102 trial did show some normalization of EEG signals in children treated with the ASO, said Elizabeth Berry-Kravis, professor of pediatrics, neurological sciences, and anatomy and cell biology at Rush University Medical Center in Chicago, Illinois, who is collaborating with the company on the trial. But the team did not see the same effect when they restarted the trial at lower doses.
As the community waits for results, families are watching closely. On Tuesday, when the family portion of the conference began, the conference halls filled with families and caregivers of people with Angelman syndrome.
These rooms were significantly larger than those for the research presentations, and talks were often punctuated by shouts — how some people with Angelman syndrome, or ‘angels,’ as many within the community refer to them, best communicate. The meeting is one of the few places where caregivers don’t feel the need to explain or apologize for their child’s differences, a mother told me. At a presentation on industry updates, a toddler who was recently diagnosed with Angelman syndrome played in the back of the room, his father watching over him.
Five companies with therapeutics under development took the stage to present data. Ultragenyx reported the same positive interim findings of GTX-102 it had described the day before. By giving the information to families, the company hopes to alleviate any remaining concerns in the community about the FDA pause, Berry-Kravis told me at the conference.
Roche is continuing its clinical trial of its ASO, Rugonersen, a representative told the audience. And Ionis launched a clinical trial of its own ASO, ION582, in December 2021. Other treatments under development include a gene therapy, still in the preclinical stage, from PTC Therapeutics, based in South Plainfield, New Jersey; and an oral drug from Australian company Neuren Pharmaceuticals that aims to correct synaptic function and is entering a phase 2 trial.
The presentations were followed by an expert panel discussion, during which the moderator asked the panelists what they believe to be most important for the success of a therapeutic. Ina Brünig-Traebert, global program team director at Roche, said it’s managing expectation bias and not jumping to conclusions prematurely — both done to “protect” the studies. As a practice, many companies, including Roche and Ionis, do not share interim data about their trials, out of concerns for influencing later results. None of the ASOs have undergone a placebo-controlled trial, and the trials can easily be biased — even by too much discussion on social media, said Rebecca Crean, executive director of clinical development at Ionis. As Brünig-Traebert put it in her presentation, “The bigger the placebo effect, the more difficult it is to detect a drug effect.”
That protection has to be balanced, researchers and clinicians agree, with the desire to give families hope. That’s partially what the conference is about: keeping companies and researchers in touch with the families, while also helping the families see what might be coming. That night, the resort staff reserved the entire pool — massive water slides included — for a conference pool party. After presentations on Wednesday, parents coaxed their children to pose for photos with the clinicians who specialize in their rare condition.
A
t the conference’s Wednesday evening reception, actors dressed as mermaids with full tails were pulled into the meeting room on wagons and set before an ocean-themed backdrop as entertainment for the children. Burdine watched as a young girl with Angelman syndrome sat cross-legged before them, mesmerized.Burdine again thought about her daughter. Sophie, like many children with Angelman, communicates with an iPad. One day a few years ago, she was screaming and no one knew what was wrong. Sophie used the iPad to type “doctor, hospital, arm, cast.” Burdine didn’t even know that Sophie knew the word “cast.” An X-ray revealed a broken bone at the back of Sophie’s arm. Burdine looked at the children moving around the reception, at the promise they held, and reminded herself, “That’s Sophie, too. These are her peers.”

John and Ronda Casner were also at the reception. They are grandparents to an ‘angel’ and have followed the research for more than 15 years. Ronda Casner remembers when the Angelman model mice were first announced and when researchers were still struggling to unsilence the paternal copy of UBE3A. Particularly exciting is the idea of being able to diagnose a child early and, one day, boost UBE3A early enough to weaken the syndrome, “instead of just trying to manage the disability,” John Casner said.
Still, in back of the researchers’ minds is the fact that failure could be around any corner. As promising as initial trials seemed, gaboxadol ended up working for adults but not for young children — because, as Levin and his colleagues later discovered, the drug acts on a receptor that changes its expression in adolescence. Likewise, trials “can tank any time because of a side effect,” Berry-Kravis said, adding that Ultragenyx, Roche and Ionis have each selected a slightly different ASO formulation for their treatment, and although those differences may prove negligible, it is also possible that a small change in the molecule could lead to harmful side effects in one ASO and not another.
These new drug modalities represent real hope, and a possibility of doing more than just ‘managing disability,’ as John Casner mentioned. It also means the field might need to grapple with what success means for different types of treatments. For some, small improvements on the CGI — a better night’s sleep and better coordination, for example — might represent change that meaningfully benefits a child’s quality of life. That may be enough of an effect for a drug taken in liquid form, such as the one Neuren has under development.
But an ASO that requires monthly lumbar punctures, or a gene therapy that has permanent effects, should have a different bar, Levin told me. Those drugs carry a higher risk and therefore must show greater benefit.

E
arlier this month, Ovid threw a grand opening party for its offices in the new Hudson Commons building in New York City. Levin, his colleagues and some families who have participated in Ovid’s trials gathered on a 14th floor terrace, skyscrapers all around them. Leaders from the New York City biotech scene and a representative from the Manhattan borough president’s office toasted the company’s future.Ovid is doubling down on Angelman syndrome and other rare genetic neurological conditions. Since stopping the gaboxadol trial, the company has launched a new approach for Angelman, one that is even more experimental than ASOs — using what is called a short hairpin RNA to unsilence paternal UBE3A. That work is in preclinical trials, and it will be years before the researchers have results to share. When Ovid started its first Angelman syndrome trial, it was the only company in the ring. Since then, multiple companies have stepped in, and there are now more than 20 therapeutics in the works.
As the field progresses, Levin and his colleagues are also hoping to understand how people with Angelman syndrome respond to placebo, and how the condition affects people over time. To that end, last month Ovid released the data from its placebo-controlled gaboxadol trial and has encouraged other companies to do the same.
Just before the official ribbon-cutting on Ovid’s new terrace, the Manhattan skyline off to his left, Levin once again turned to the gaboxadol failure. “We’ve learned from that,” he told the crowd, “and we will succeed.”