Welcome to the August issue of Going on Trial, a monthly newsletter covering clinical trials and drug development for autism and related conditions. This month, we explore the electroencephalagraphy (EEG) biomarkers that are part of new clinical trials for dup15q and Angelman syndromes, hear a parent’s take on the risks and benefits of experimental treatments and review the initial results from an ongoing trial of a gene therapy for Rett syndrome.
Balancing beta:
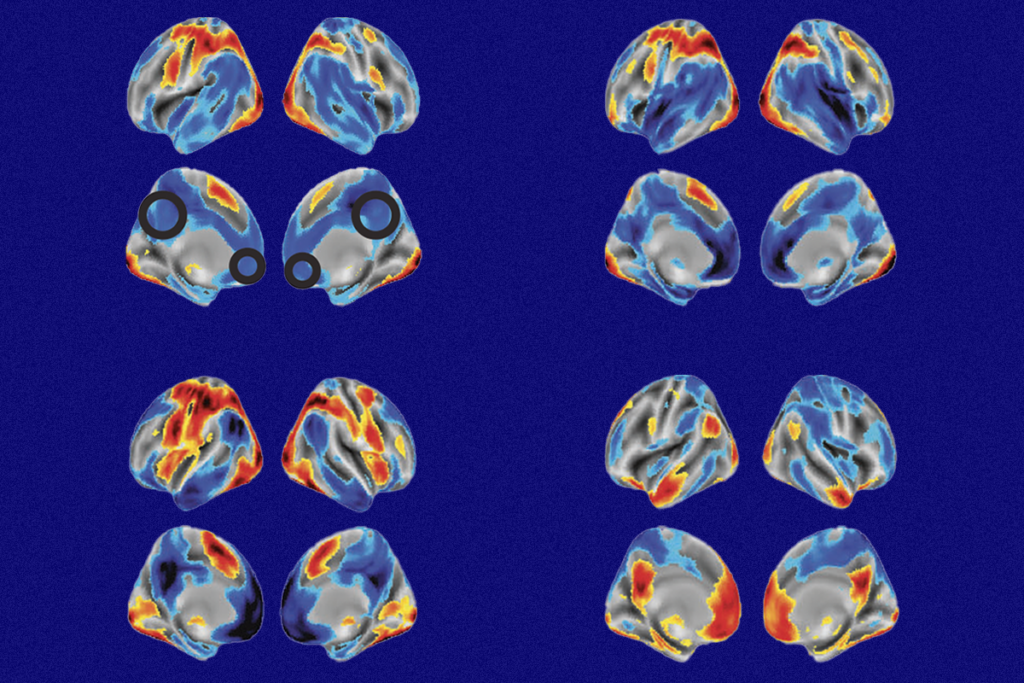
An EEG biomarker could reveal whether a potential treatment for dup15q syndrome, an autism-linked condition, has reached its intended molecular target in the brain, according to a presentation last month at the 2023 Dup15q Alliance/Angelman Syndrome Foundation Science Symposium.
People with dup15q have heightened EEG activity in the beta frequency band (12 to 30 Hz). “It’s almost unprecedented, the robustness of this dup15q biomarker. You can see it with the naked eye,” says Shafali Jeste, chief of neurology at Children’s Hospital Los Angeles. “It’s not subtle.”
Beta band activity is known to increase in people taking benzodiazepenes, which boost gamma-aminobutyric acid (GABA) signaling. Jeste and her colleagues hypothesize that aberrant GABA signaling could explain the EEG signature seen in people with dup15q syndrome.
People with the syndrome carry extra copies of the 15q11.2-q13.1 chromosomal region, which contains three genes that encode subunits of the type A receptor for GABA, including the alpha-5 subunit. This may lead to an overproduction of alpha-5-containing GABAA receptors, which would increase inhibitory signaling.
It is not yet known if increased GABA signaling is responsible for the syndrome’s traits, says Larry Reiter, professor of neurology at the University of Tennessee in Memphis, whose team first spotted the beta signature more than 10 years ago.
Jeste and other researchers are testing the utility of this biomarker in a phase 2 trial of basmisanil, an experimental drug that selectively blocks GABAA-alpha-5 receptors. The team hypothesizes that the drug could reduce excess GABA signaling, decrease beta band power and improve adaptive behaviors and everyday functioning in people with dup15q syndrome, says Joerg Hipp, biomarker and experimental medicine leader at the drug company Hoffmann-La Roche, which sponsors the trial. And if it does, it would provide support for the idea that increased GABA signaling contributes to syndrome traits, he adds.
Beta band power is also being tested as a proof-of-mechanism biomarker in an Angelman syndrome trial — but in the opposite direction. People with Angelman syndrome have mutations or deletions, rather than duplications, in the 15q11.2-q13.1 chromosomal region. Those with a deletion have lower beta band power, which may be driven by a reduced number of GABAA-alpha-5 receptors.
The phase 2 trial, also sponsored by Roche, is evaluating alogabat, an activator of GABAA-alpha-5 receptors, in children and teenagers with the Angelman syndrome deletion genotype. The drug has previously been tested in autistic adults. After adjusting dose levels in the first part of the study, the researchers plan to measure brain activity using EEG to determine whether the treatment increases beta band power as hypothesized.
Weighing risks:
In 2021, an experimental drug trial for a rare form of epilepsy ended in tragedy. The two 2-year-olds taking the drug — Valeria Schenkel and Lucy Greenblott — developed hydrocephalus, a buildup of fluid in the brain, and Valeria died as a result, according to The New York Times.
Despite this grave setback, there is still reason to hope, says Justin West, president and director of clinical medicine at the KCNT1 Epilepsy Foundation and father of Andrew, a 6-year-old boy with the condition.
Before the two girls developed hydrocephalus, their seizures reduced and their development progressed. This is a sign that the condition is treatable, and perhaps even curable, West says.
Spectrum spoke with West about why he thinks it’s important to continue developing therapies for KCNT1-related epilepsy and other rare forms of epilepsy, despite the risks.
Spectrum: What’s daily life like for Andrew?
Justin West: He’s 6 years old, but functionally he’s 6 months old. He can sit; he can go from lying down to sitting up; he can reach for his bottle; and he can drink on his own. We use a wheelchair to move him around, and he needs a power lifter to get in and out of the bath. We spoon-feed him; he’s totally dependent on us.
And that’s what his life will always be like, unless we find him a drug to change his trajectory.
S: As a parent, how do you weigh the potential risks and benefits of an experimental treatment?
JW: In this community, the way we perceive risk is very different than the way a scientist or drug developer would perceive it — or, quite frankly, than the way our epilepsy specialist would perceive it. They’re petrified of hurting our children, but our children have already been traumatically injured by this condition.
And so those of us who are raising these children, I think we have a pretty high tolerance for things to go wrong. Because things have already gone so wrong. I always tell my doctors, “You can’t hurt my kid any more than he’s already been hurt.”
Read the full interview.
Drug samples:
- It was a big month for TSHA-102, an experimental gene therapy for Rett syndrome. The biotech company Taysha Gene Therapies secured approval from an independent data monitoring committee to dose a second woman with the treatment. And the U.S. Food and Drug Administration (FDA) approved Taysha’s investigational new drug application to test the treatment in children (the company’s original trial involves adults and is based in Canada). The company also released early results from the first recipient: TSHA-102 was well tolerated with no serious adverse events six weeks after treatment. At an evaluation four weeks post-treatment, the participant showed improvement in breathing patterns, sleep quality, vocalizations and motor skills. She was able to sit unassisted for the first time in a decade and to hold objects for the first time since infancy, according to a company press release.
- Anavex Life Sciences has a tendency to change the endpoints in its trials for blarcamesine — a Rett syndrome drug — after the trial has ended, making the drug appear more effective than it otherwise would, writes STAT reporter Adam Feuerstein in a column. But Anavex must use the same endpoints that Acadia Pharmaceuticals used to obtain FDA approval of trofinetide for Rett earlier this year, Feuerstein writes: The approved drug “is like Wonder Woman’s lasso of truth,” and “deviating from the FDA-endorsed standards will be immediately disqualifying.”
- And more Rett news: Australian pharmaceutical company Neurotech International dosed their first Rett syndrome participant with NTI164, a cannabis-derived drug. The company intends to dose 14 girls and women aged 5 to 20 and plans to share initial results from the trial at the beginning of 2024.
- The combination of the antipsychotic risperidone with the amino acid derivative l-carnitine is more effective at reducing irritability and hyperactivity in autistic children than risperidone alone, according to results from a clinical trial in Iran. However, the side effects of lethargy, stereotypic behaviors and inappropriate speech were similar in both groups.
- The cost of translating consent documents from English into other languages may explain why some racial and ethnic minority groups are underrepresented in clinical trials, Science reports. Industry-sponsored studies translated twice as many consent documents as non-industry studies, according to a study from Nature.
- On 15 August, the FDA published final guidance on informed consent for clinical trials, replacing the guidance published in 1998, reports the law firm Ropes & Gray. The update includes recommendations on when new information needs to be shared with participants and which consent form changes don’t need to be approved by an institutional review board.
That’s all for August! Make sure you subscribe so you can receive this newsletter in your inbox every month, and feel free to get in touch with feedback or story ideas: [email protected]