Mutations in the autism-linked gene ANK2 may prevent the protein product of SCN2A — another gene strongly associated with the condition — from anchoring to dendrites, according to a study published today in Neuron. The work uncovers a previously unknown interaction between the genes that could be targeted therapeutically in multiple ways.
Mice missing one copy of ANK2 and those lacking SCN2A show similar neuronal changes — including dampened excitability in dendrites, the new analysis found, suggesting the proteins may work together to regulate synaptic plasticity.
“The more we study SCN2A, the more we realize the importance of its interactions,” says co-lead investigator Kevin Bender, associate professor of neurology at the University of California, San Francisco. “It’s a keystone gene, so if we pull on the SCN2A thread, we might understand more about the actual mechanisms that underlie ASD.”
SCN2A codes for Nav1.2, a sodium ion channel enriched in dendrites. Unlike sodium channels in the axon, which propel impulses toward other neurons, Nav1.2 sends the impulse backward so that synapses can adjust their strength according to neuronal activity.
How Nav1.2 docks onto dendrites was previously unclear. Many assumed it locks onto ANKG, the scaffolding protein that tethers sodium channels in the axon. But instead, the new study found, the sodium channel anchors onto ANKB, the protein product of ANK2.
“From a basic science point of view, that’s a breakthrough,” says Vann Bennett, professor of molecular biology at Duke University, who was not involved in the work. “Myself and many others were operating under assumptions that turned out to be wrong. This clearly opens our eyes.”
The new findings also help in a larger effort to identify common functional pathways that unite many of the 100-plus genes linked with high confidence to autism. SC2NA had long seemed an outlier, but the new work suggests that — like many other autism-linked genes — it may play an important role in synaptic function.
A
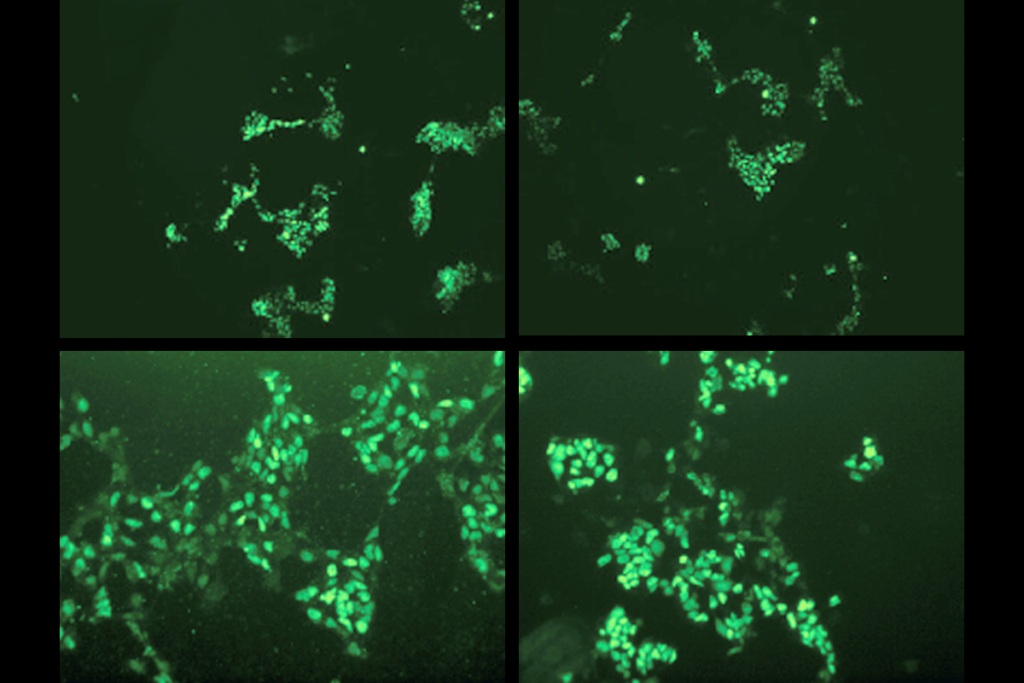
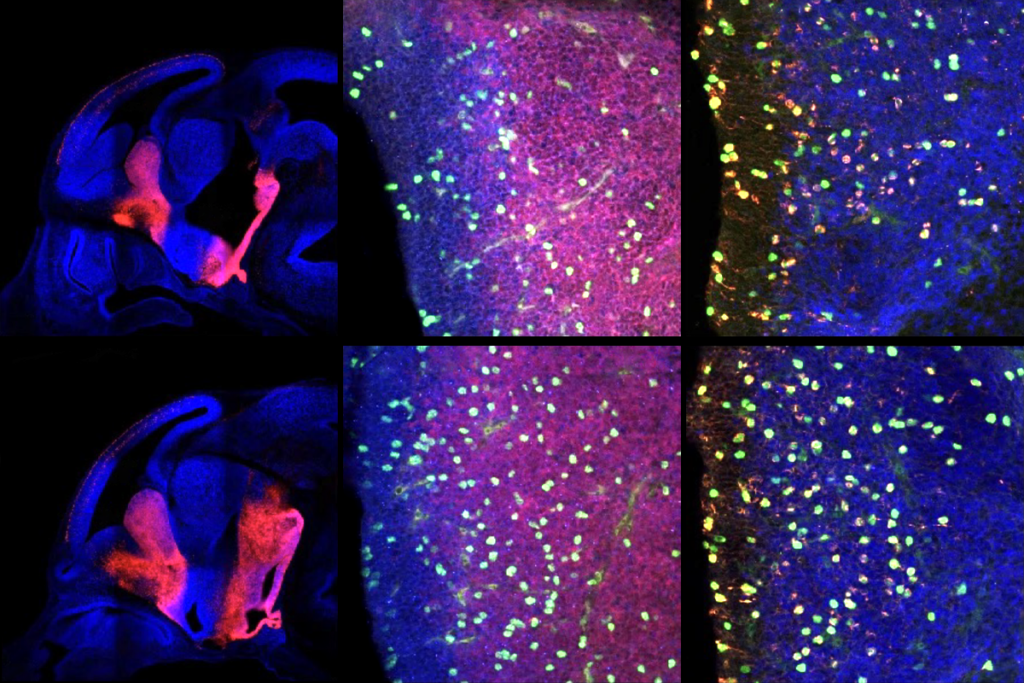
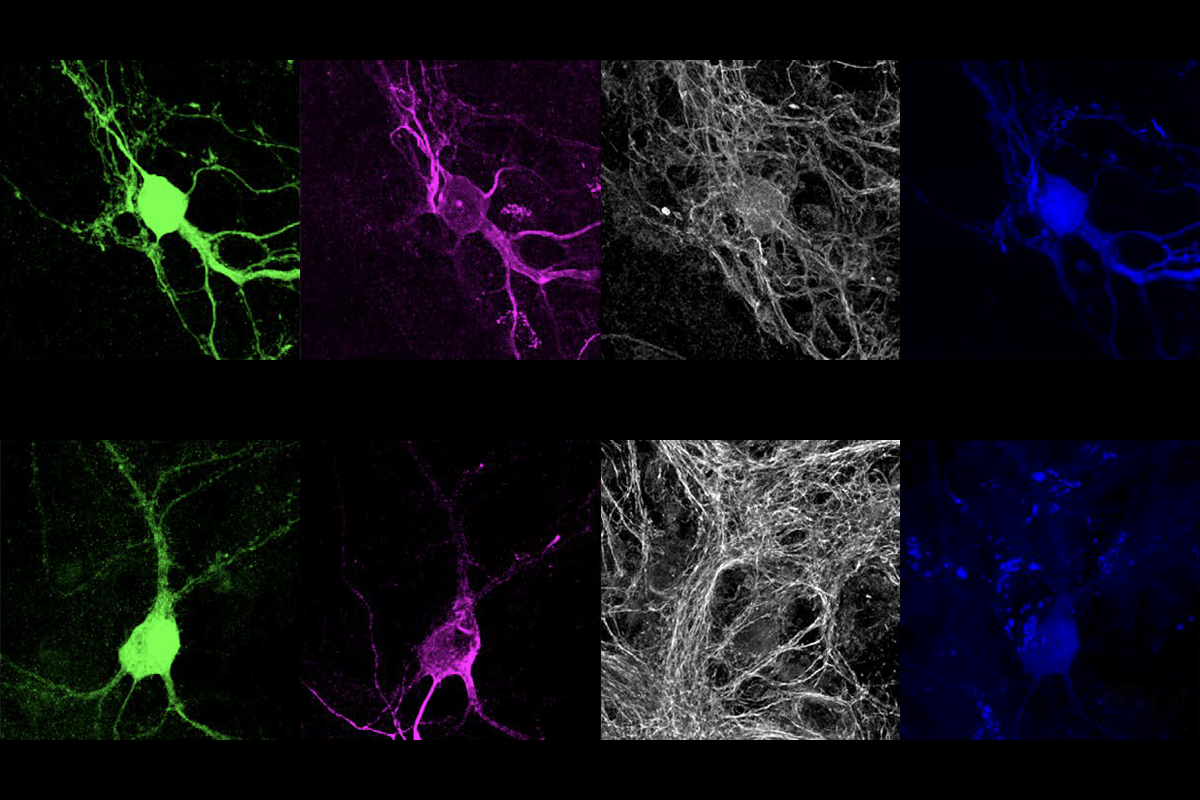
nkyrins, the protein family that includes ANKB and ANKG, are “like parking spots for the channels,” says co-lead investigator Paul Jenkins, assistant professor of pharmacology and psychiatry at the University of Michigan, but “early in development, there aren’t any parking spots” available.Antibody staining for Nav1.2 in cultured neurons revealed that the sodium channel migrates from the axon to the dendrites when mice are around 1 week old. And when the team stained for scaffold proteins, ANKB, not ANKG, appeared in the same sites of the dendrite as Nav1.2.
That relocation might not occur in people with ANK2 mutations, the team speculates. The event corresponds to a human age of 1 year old, when some children miss developmental milestones and get flagged for an autism diagnosis.
“That [timing] perfectly jibes,” Bender says.
In support of that idea, in neurons from mice that lack one working copy of ANK2, the sodium channel did not relocate to the dendrites, the study found. And those dendrites released less calcium, which indicates reduced excitability.
“It’s a really important piece of the puzzle,” says Karun Singh, senior scientist at the Krembil Research Institute, who was not involved in the work. “Dendritic excitability is not something that’s received much focus, but this may encourage others to explore that avenue.”
Recordings of neuronal activity in the mice revealed less frequent excitatory postsynaptic currents in mice lacking ANK2 than in their wildtype littermates. And those neurons formed synapses with fewer AMPA receptors, hinting that these connections are inactive.
T
aken together, the dendritic changes could dampen synaptic plasticity. Under typical conditions, a synapse strengthens when activity there produces an action potential — a process known as long-term potentiation (LTP).But neuronal activity in mice missing ANK2 doesn’t trigger LTP, the study found. This difference might be due to less Nav1.2 sending action potentials backward, so the synapse doesn’t “know” whether its activity produced an impulse, Bender and his colleagues say.
Although the study provides solid evidence that the two genes join forces to regulate dendritic activity, ANKB likely contributes to other neuronal pathways, Singh says. “I think this is just the tip of the iceberg.”
But therapeutic strategies to boost ANKB could benefit people with mutations in either gene, Jenkins adds. “If you turn up the ankyrin level at the dendritic membrane, we now have more parking spots for the channel to hang out in. Even SCN2A haploinsufficiency could be treated by recruiting more channels.”
Another approach might be to stabilize the interaction using small molecules. In collaboration with other groups, the researchers are using machine learning to try to identify such molecules.
They say they also hope to uncover how ion channel tethering is regulated. So far, it’s unclear how the ankyrin family — with nearly identical binding sites — distinguishes among channels in different neuronal compartments.