This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
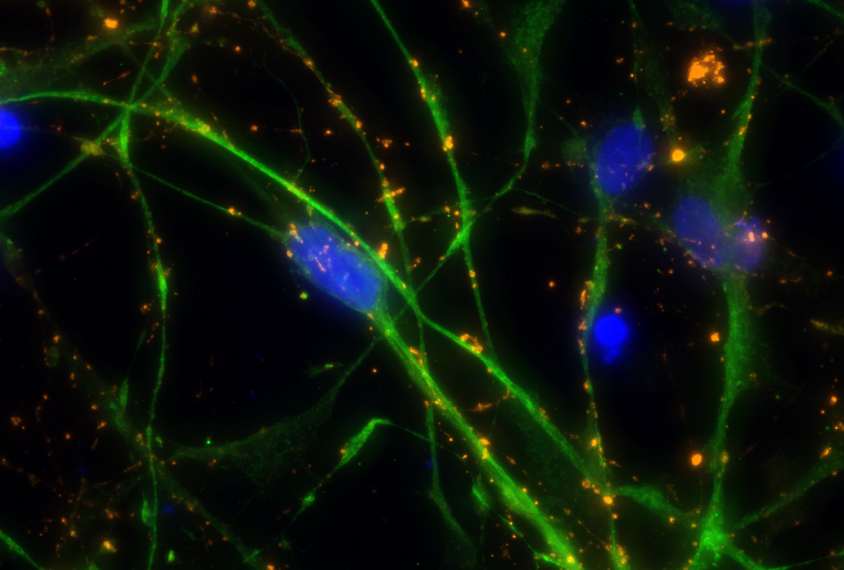
Cells derived from the dental pulp of children with Williams syndrome hint at a treatment approach for autism.
Cells derived from the dental pulp of children with Williams syndrome divide rapidly and often die before maturing into neurons. The finding offers clues about the biology underlying the syndrome, which is marked by an intense interest in social interactions.
Children with Williams syndrome are missing a region of chromosome 7 called 7q11.23. They show some features of autism, such as language and cognitive difficulties. But they are unusually social, suggesting that a gene or set of genes on chromosome 7 plays a role in social interest.
The new study, published 18 August in Nature, homes in on a gene in the 7q11.23 region called FZD9, which is needed for neurons to mature1. Missing the gene may alter the number of neurons in the brain and change how they communicate with each other.
“We think that by just having this defect, you can increase cell death and enhance the connections between the neurons that survive,” says lead researcher Alysson Muotri, associate professor of pediatrics and cellular molecular medicine at the University of California, San Diego.
Muotri says the enhanced connections between surviving neurons may drive social interest in children with Williams syndrome. “We can take advantage of that pathway to modulate the social behavior in autism,” he says.
Muotri’s team has tapped teeth for clues about autism before. In 2014, the researchers used dental pulp cells from a boy with the condition and uncovered a mutation in a gene that may help to shape synapses, or neuronal connections.
For the new study, they collected discarded baby teeth from five children with Williams syndrome and four typically developing children. Four of the children with Williams syndrome are missing the entire 7q11.23 region.
The fifth child with Williams has a partial deletion, which usually produces subtler social and cognitive features than the full deletion does. This child scored slightly better on a cognitive test and is less social than the other four children with Williams syndrome.
The researchers extracted cells from the dental pulp and induced them to form stem cells. They then transformed the stem cells into neural progenitor cells, which give rise to mature neurons.
Cells from children missing the entire 7q11.23 region divide too rapidly, the researchers found. These cells are more likely to die before maturing into neurons than are cells from either the controls or the child with a partial deletion.
This cell death may explain reports of low brain volume in children with Williams syndrome, the researchers say2. They verified these reports themselves by scanning the brains of 19 people with the condition, and found a smaller cortical surface area than in age-matched controls.
A small fraction of cells from children missing the entire 7q11.23 region do mature into neurons. But these neurons have unusually long signal-receiving branches, called dendrites, and form too many connections with other neurons.
The researchers saw a similar increase in dendrites and synapses in postmortem brain tissue from two people with Williams syndrome. “I see this as evidence that our cellular system is recapitulating human development in a dish,” Muotri says.
The researchers used electrodes to record the electrical chatter between the neurons. The irregular cells forge networks that produce intense bursts of electricity, compared with the steady stream of pulses emanating from networks of control cells.
“The idea of changes in circuitry has long been advocated in the Williams syndrome research field, and this work strongly supports this theory,” says Lucy Osborne, professor of medical genetics at the University of Toronto, who was not involved in the study.
Neurons derived from the child with a partial deletion resemble those from controls. This led the researchers to focus on FZD9, which is missing in children with the full deletion but intact in the child with a partial deletion. FZD9 is a receptor for a protein involved in the WNT signaling pathway, which has been linked to autism.
Blocking FZD9 expression in control cells and cells with only a partial deletion mimics the full deletion’s effects on dendrites and synapses, the researchers found. Activating the WNT pathway reverses this effect.
The researchers plan to test the behavioral effects of blocking FZD9 in mice. If it boosts social interest, it could represent a treatment option for autism. They also plan to study other genes that are missing only in cells with the full deletion.