A potential new gene therapy for Dravet syndrome increases survival and prevents seizures, according to initial tests in mice, a new study shows. The approach delivers two viruses—each carrying a portion of the therapeutic gene—selectively to inhibitory interneurons, the cell type affected in the condition.
“This therapy has the potential to provide a long-term, possibly one-time treatment,” says Debopam Samanta, professor of pediatrics at the University of Arkansas, who was not involved in the work.
SCN1A, the gene altered in most people with Dravet syndrome, encodes a sodium ion channel that is essential for interneurons to fire and transmit signals. About 40 percent of people with the condition also have autism. Because SCN1A is too bulky to squeeze into an adeno-associated virus (AAV), which is commonly used to deliver gene therapies, previous approaches have instead delivered a smaller artificial transcription factor or antisense oligonucleotides (ASOs) that prompt expression of endogenous SCN1A.
These approaches turn up expression of both copies of the gene, including the dysfunctional allele, a strategy that might benefit people who have variants that dampen expression of SCN1A. But for people whose variants give rise to an altered form of the sodium channel, amplifying expression could make matters worse, explains study investigator John Mich, senior scientist at the Allen Institute for Brain Science.
Instead, Mich and his colleagues aimed to provide a fresh copy of SCN1A. They split the gene in two and packaged each into a separate virus. Certain cells then translate the two halves and fuse them to form the complete protein. In theory, this type of therapy could be given to everyone with SCN1A-linked Dravet syndrome, regardless of their genetic variant, Mich says.
And by switching on expression only in affected cells—a subtype of inhibitory interneuron—the dual virus method is less toxic and more effective than non-selective approaches, the study found. The work was published in Science Translational Medicine in March.
A
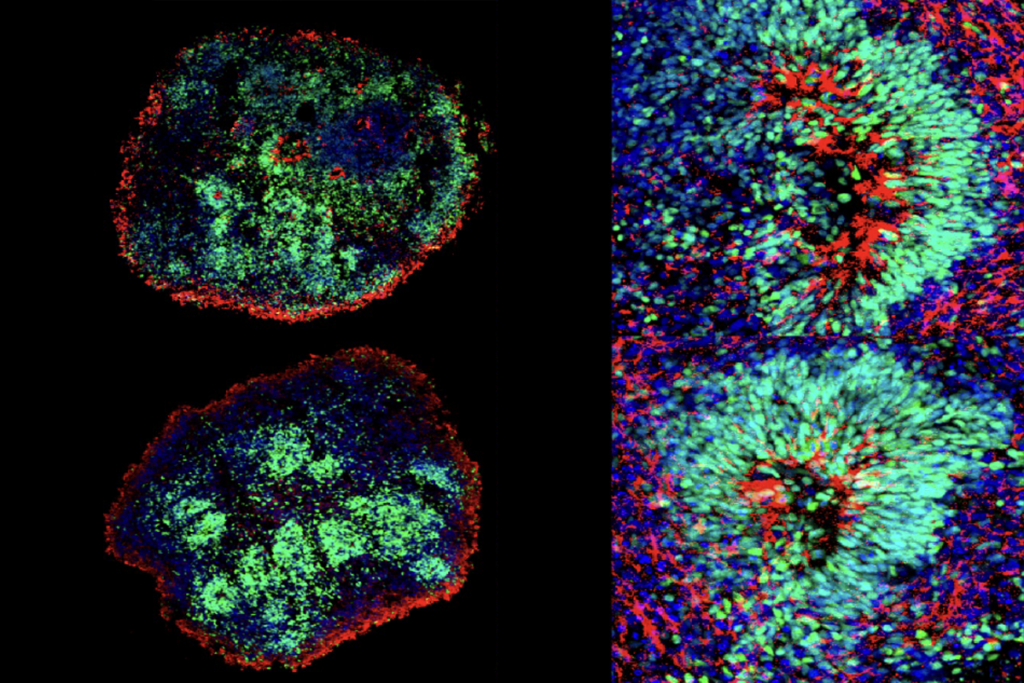
lthough the sodium channel is expressed in excitatory and inhibitory neurons, only the latter are affected by loss of SCN1A function. Deleting one copy of the gene in the excitatory cells of mice has no noticeable effect, according to a 2013 study.But SCN1A haploinsufficiency in inhibitory neurons in mice leads to hallmarks of Dravet syndrome, including spontaneous and heat-induced seizures, as well as cognitive and behavioral problems.
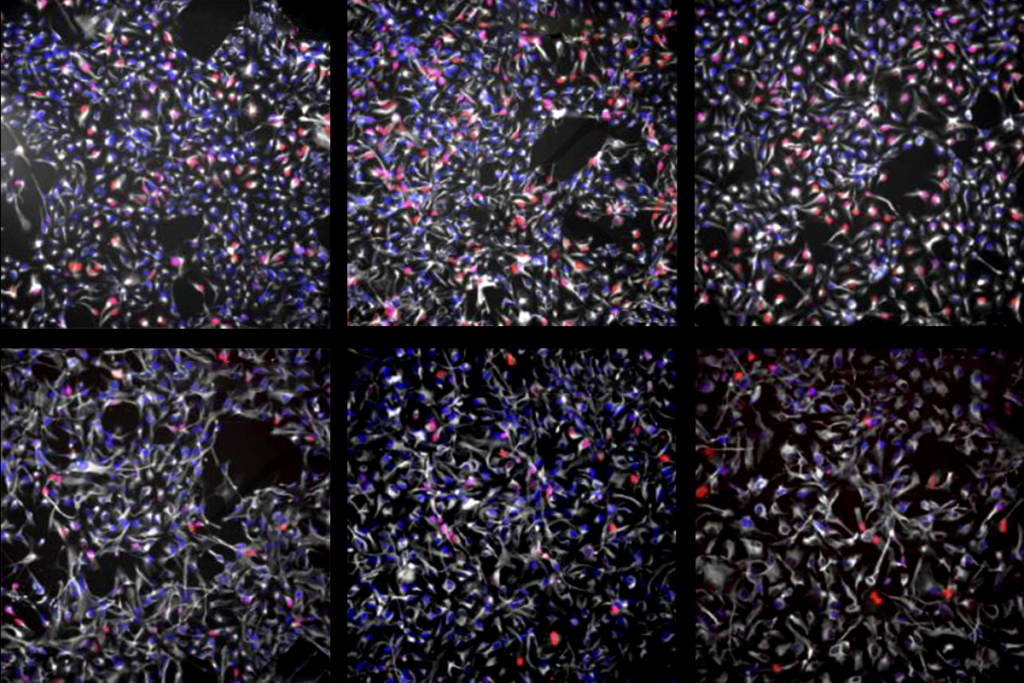
Mich and his colleagues targeted inhibitory interneurons using AAVs that have an enhancer that drives expression in telencephalic GABAergic interneurons.
To express the full-length sodium channel, the scientists used a method of protein splicing that has proved effective in replacing the lengthy gene responsible for Duchenne muscular dystrophy. To the two halves of SCN1A, the group added protein segments called inteins. After translation of SCN1A, the intein fragments connect and are then spontaneously removed, knitting the two halves of SCN1A together in the process.
Half of mice missing one copy of SCN1A die from seizures within a few months of birth. But injecting the dual virus into the animal’s brain completely prevents premature death, the study found.
Dravet model mice—like people with the condition—are prone to heat-induced seizures, which are sometimes fatal. (Children with Dravet syndrome can’t play outside on warm days without risking a seizure.) When the researchers bumped up the body temperature of treated mice, they all appeared to remain healthy, whereas more than half of the untreated rodents showed seizures. The dual AAV also reduced spontaneous seizures, electrode recordings revealed.
Injecting the viruses into another mouse model of Dravet syndrome also enhanced survival and prevented seizures, the study found.
But the AAV’s effectiveness depends on its cellular selectivity: When the team swapped the enhancer for a sequence that drives expression in all neurons, the treatment only partially protected mice against seizures and premature death.
D
espite these promising results, there are problems to solve before the viruses can be given to people.For instance, doubling the viral particles needed to express SCN1A “may require a particularly high dose, which can pose significant risks,” says Moran Rubinstein, associate professor of human molecular genetics and biochemistry at Tel Aviv University, who was not involved in the work.
Indeed, last year a young girl died after receiving a high dose of AAVs as part of a clinical trial for Rett syndrome.
But future research to establish the minimum therapeutic dose, and targeting of specific brain regions, could limit the amount of virus needed, says the study’s principal investigator, Boaz Levi, associate investigator at the Allen Institute for Brain Science.
Provided the dual AAVs can be delivered in safe quantities, they might one day be given to children with Dravet syndrome in combination with ASOs and other approaches, as is currently happening in children with spinal muscular atrophy, Levi says. “The nice thing is that these therapies work on orthogonal strategies” and could complement each other, he adds.