A drug containing snippets of RNA reverses defects seen in neurons derived from children with Timothy syndrome, a study published today in Nature shows. The compound, tested in rats that contain transplanted human neurons, could enter clinical testing as early as next year, the study investigators say.
Timothy syndrome is a rare, life-threatening genetic condition characterized by heart problems, seizures and autism. It stems from a mutation in the gene CACNA1C, which encodes a critical calcium channel. Neurons with faulty calcium signaling have shortened dendrites, show atypical firing and fail to form critical brain circuits during development.
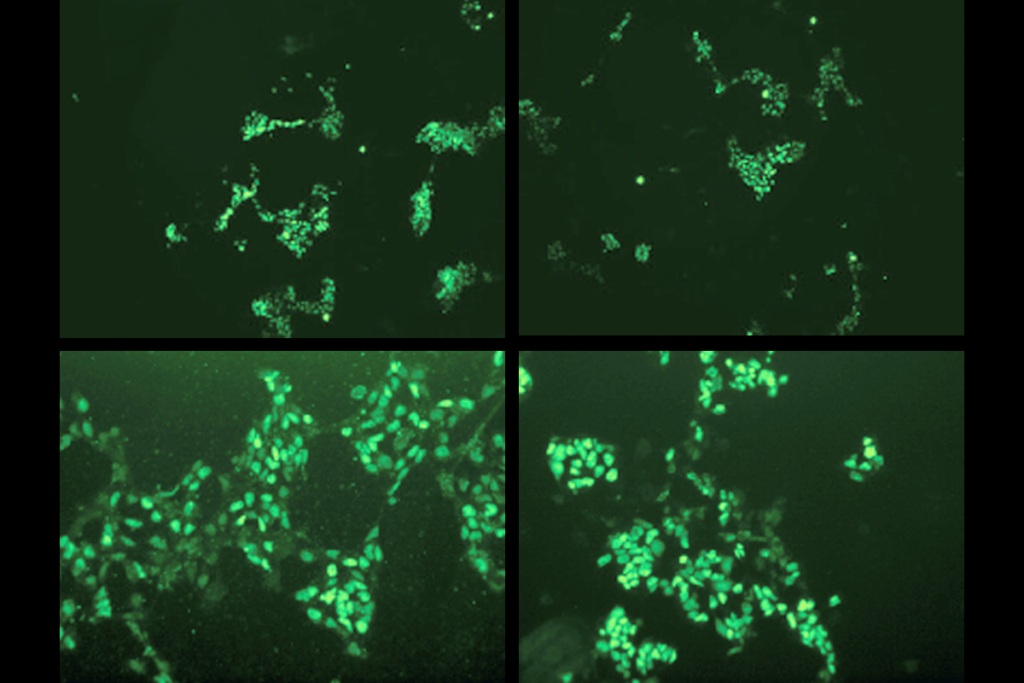
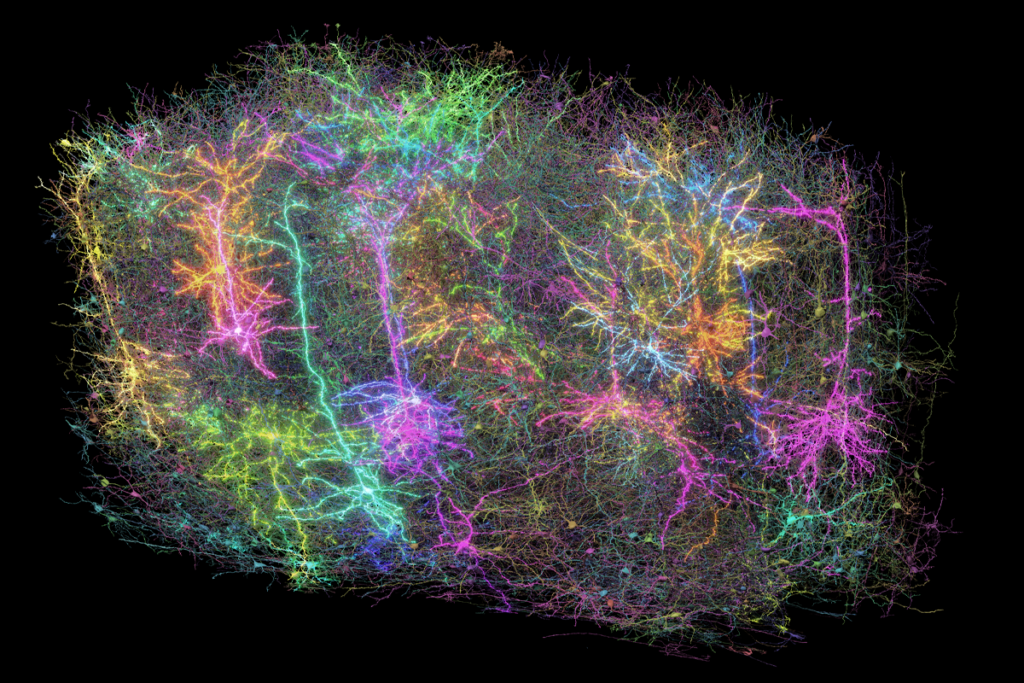
Clumps of neurons made using stem cells from children with Timothy syndrome show atypical calcium uptake and mimic all of these defects, previous research shows. But the altered dendrites, firing patterns and circuit development can be fixed using antisense oligonucleotides (ASO)—short strands of RNA that complement a target sequence of DNA—to mute the CACNA1C mutation.
“[The ASOs] go in, interfere with the process, and within a few days, the RNA that carries the mutation is essentially gone,” says lead researcher Sergiu Pasca, professor of psychiatry and behavioral sciences at Stanford University. “And that turns out to restore essentially almost all the defects that we’ve described over the past 15 years.”
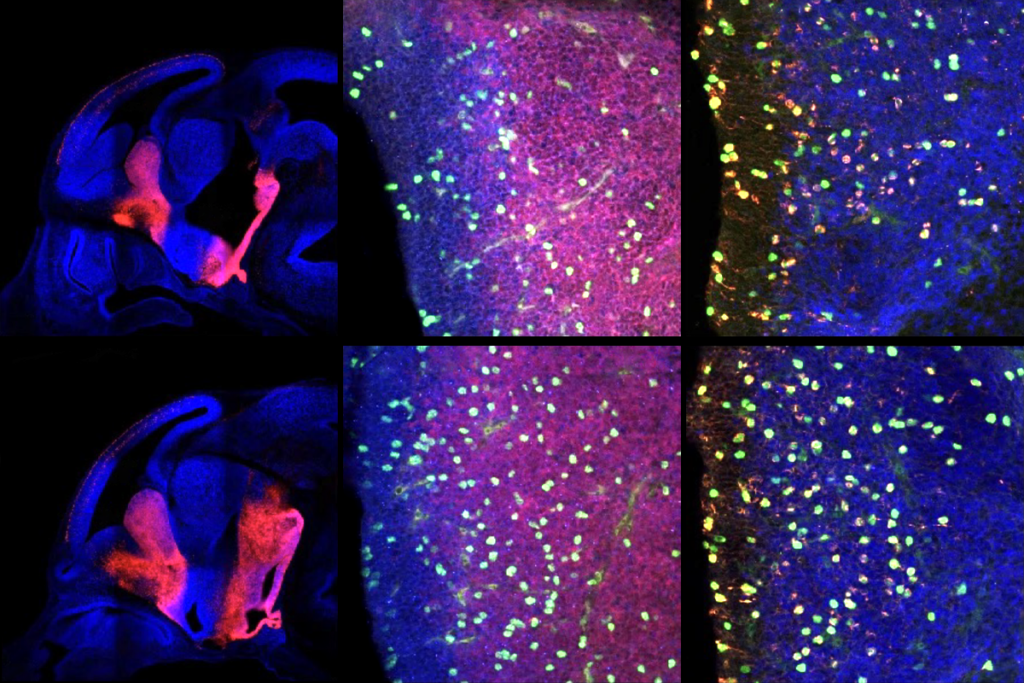
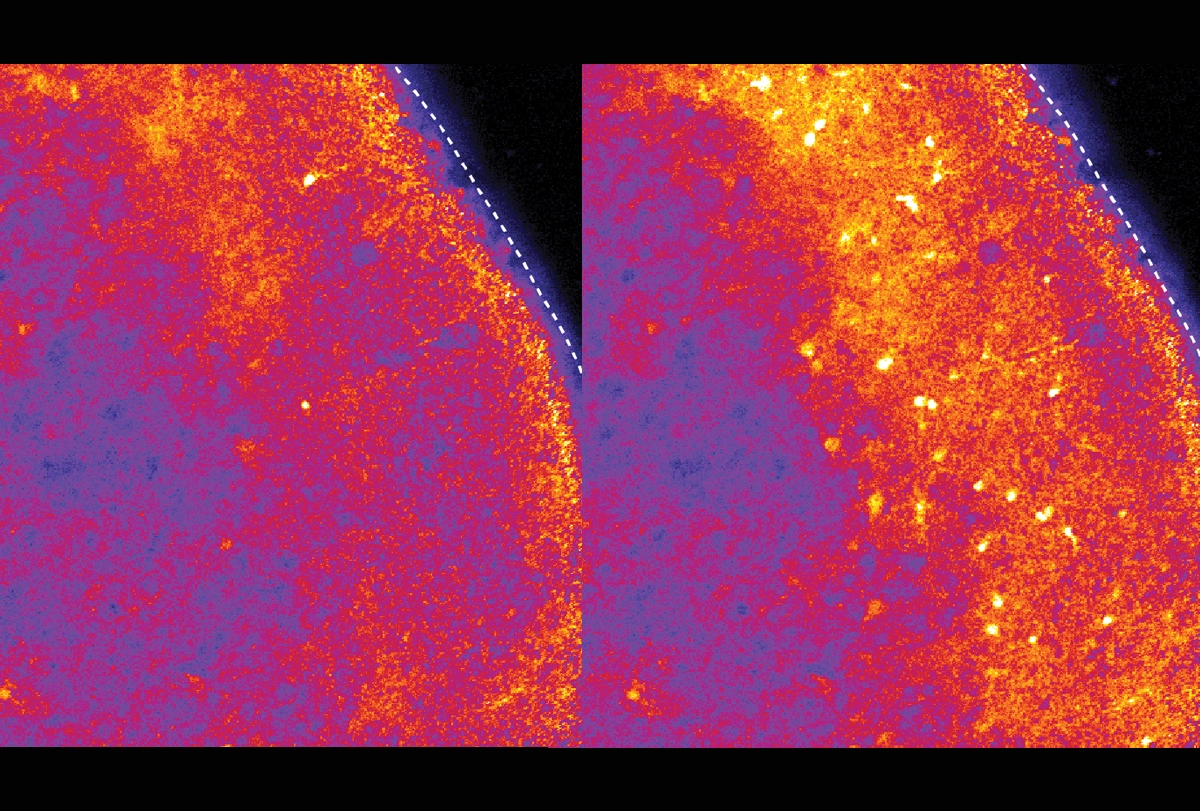
In the new study, Pasca and his team transplanted Timothy syndrome organoids into rat brains and then injected ASOs into the animals’ cerebrospinal fluid. After 7 to 10 days, the neurons showed normal dendrites and calcium uptake.
“I think this approach holds great promise for potential therapeutic approaches for neurological disorders,” says Silvia Velasco, who leads the neural stem cells research group at the Murdoch Children’s Research Institute. Velasco was not involved in the study but wrote a News and Views article about it for the same issue of Nature.
The approach, however, is invasive and would likely need to be repeated every few months in children with the syndrome, she adds. “I think that’s an important limitation.”
T
he new study builds on work Pasca started in 2009, when he first derived neurons from the skin cells of a child with Timothy syndrome. The team later cultured the cells in organoids and assembloids, which merge organoids to recapitulate different brain regions.The mutations that cause Timothy syndrome reside in a coding region of CACNA1C known as exon 8, which has two versions, or isoforms: one that is active during development and another that takes over after brain circuits have formed. In Timothy syndrome, the mutant isoform is active during development. So Pasca and his team set out to find an ASO that would drive the expression of its healthy counterpart.
“We’re not correcting the mutation at the DNA level, but we’re actually leveraging a normal physiological process—biasing it towards the [unaffected exon] so we move away from the pathophysiological disease,” Pasca says. “And it worked. It worked amazingly.”
The ASO worked in assembloids, but delivering it to the brain of a living animal presented new challenges. Because animal models don’t capture the defects seen in Timothy syndrome neurons, Pasca and his team transplanted their organoids into rat brains. There, the human neurons integrated with the rat neurons and continued to show the defects observed in cell culture.
The human-rat chimeras enabled the researchers to demonstrate the effects of the ASOs when delivered via injection into the fluid that swaths the brain.
T
he heart problems associated with Timothy syndrome usually lead to its diagnosis soon after birth. This timing presents an opportunity for early intervention—perhaps not early enough to address the problems with neuron migration and circuit formation, but in time to ease some of the neurological features of the condition, such as seizures, says Hongjun Song, professor of neuroscience at the University of Pennsylvania’s Perelman School of Medicine.“This is really an evolution of work,” Song says. “They are now showing that you actually can use an in-vivo model to study human disorders and test treatment strategies.”
In particular, the study has implications for other conditions that involve alternative splicing. It “provides a road map for characterizing exon splicing variation in a gene and its effect on neuronal structure and function using human neurons,” says Mustafa Sahin, director of Boston Children’s Hospital’s Translational Neuroscience Center and professor of neurology at Harvard Medical School. “Of course, there are still several hurdles to cross until such a treatment approach shows safety and efficacy in a clinical trial. Nonetheless, it’s an exciting beginning based on rigorous basic neurobiology.”
A different research team is already testing ASOs in children with Dravet syndrome, another neurological condition marked by seizures and autism. The approach similarly silences the mutant exon and boosts translation of a healthy exon.
Pasca says he and his colleagues plan to test the safety of the Timothy syndrome ASO approach in nonhuman primates and then start a clinical trial at Stanford.