This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
Studies of Rett syndrome hint at genes, cells and brain circuits that may be involved in autism — and may pave the way to treatments for both conditions.
Austrian pediatrician Andreas Rett first recognized the syndrome that would later bear his name in the mid-1960s. The first description in English, published in 1983, detailed a “progressive syndrome of autism” and other traits in 35 girls.
Rett syndrome usually arises from mutations in the MECP2 gene on the X chromosome. Children who have it — virtually all of them girls — become withdrawn, develop repetitive hand movements and often lose the ability to speak and walk. The mutations are usually fatal in boys soon after birth, but a few survivors have mild mutations or atypical forms of the syndrome.
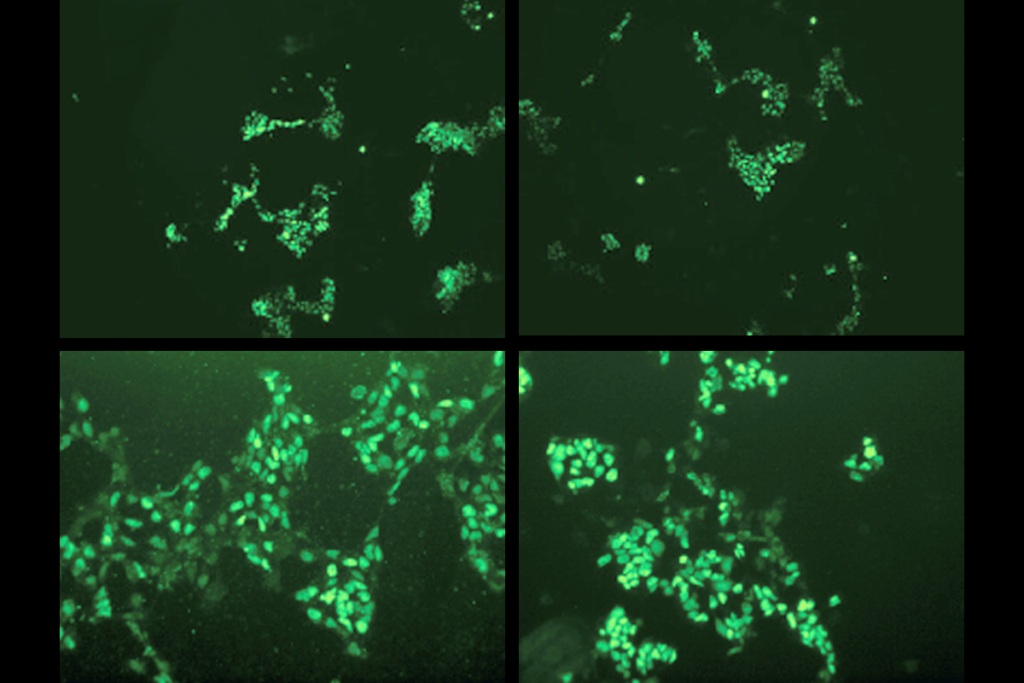
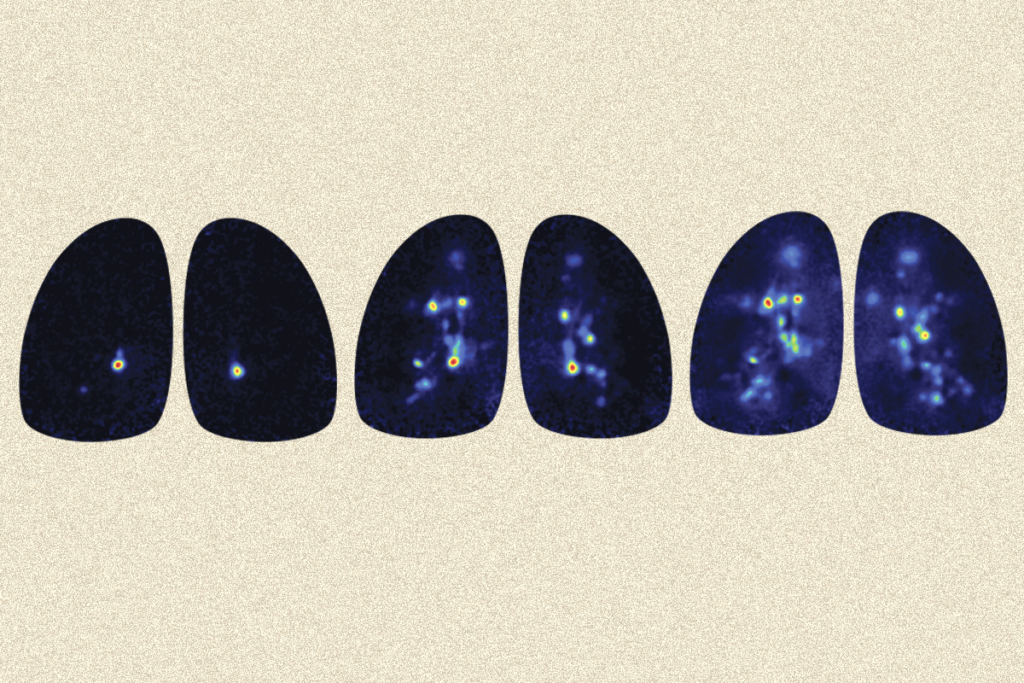
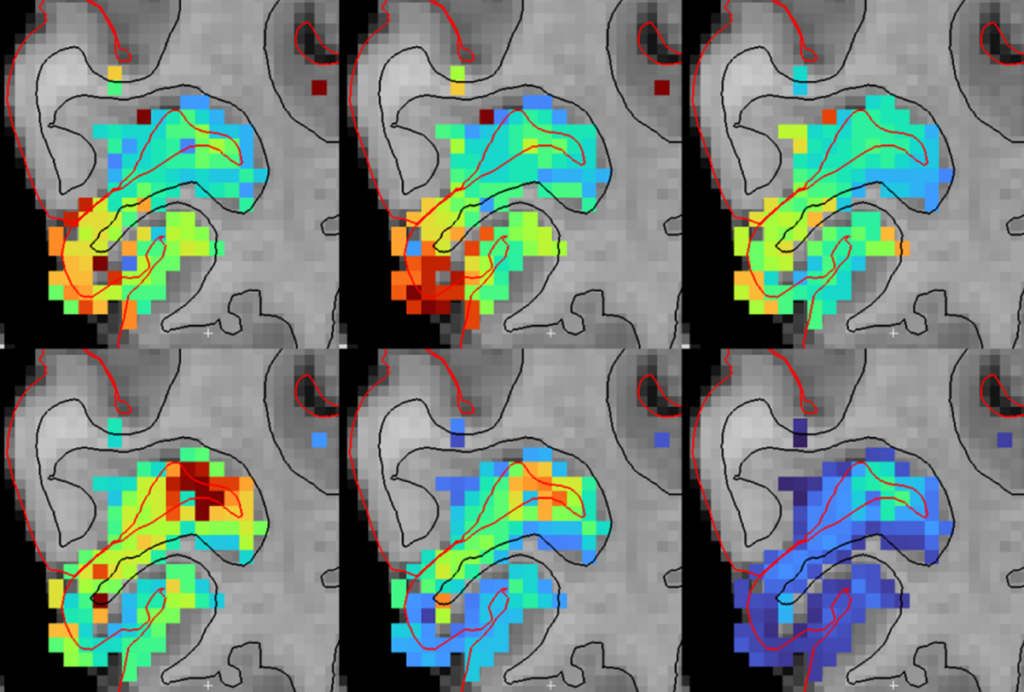
Mouse models of Rett syndrome are yielding insights into the brain cells and circuits that may be involved in the syndrome, and also in autism. Studies of Rett may even point to autism treatments.
What do Rett syndrome and autism have in common?
Quite a lot. Like autism, Rett syndrome is not apparent at birth; in both conditions, a period of apparently typical development precedes the onset of early signs.
Those signs, as well as their timing, are similar for both conditions. Between the ages of 6 and 18 months, children with Rett syndrome withdraw from social interaction and lose the ability to speak. At the same age in autistic children, parents and caregivers may notice a lack of social interest as well as communication problems.
Regression, the loss of previously acquired skills, is a hallmark of Rett syndrome. At least one in five children with autism also experiences regression. The regression happens at about the same time in both conditions and involves similar trends: loss of language and social skills.
Repetitive behaviors are common in both conditions, too. In Rett syndrome, repetitive hand movements — usually hand-wringing or touching the hands to the mouth — are often so frequent they prevent the children from using their hands in a purposeful way. The repetitive behaviors associated with autism are more varied and may include spinning, body-rocking and grinding of teeth. Autistic people also often show cognitive forms of these behaviors: routines, rituals or an intense focus on a specific interest.
Other features, such as anxiety and seizures, are also common in both conditions.
Are there differences between the two conditions?
Yes, and some are significant. For example, atypical social behavior is a defining characteristic of autism. But in people with Rett syndrome, the loss of social interest is often temporary: Over time, many girls with Rett syndrome become socially engaged again. And whereas many people with autism avoid eye contact, those with Rett syndrome often learn to use eye movements to communicate their wishes.
Movement problems in people with Rett syndrome tend to be much more severe than those in autistic people. People with autism may have poor coordination or an awkward gait. But many girls with Rett syndrome are unable to walk, and as they get older they may develop rigidity or tremors.
Rett syndrome also involves problems with the autonomic nervous system that may lead to fatal breathing abnormalities, a problem not seen in autistic people.
What can studies of Rett syndrome tell us about autism?
Scientists are investigating how regression unfolds in Rett syndrome in hopes of better understanding regression in autism. One hitch is that Rett mouse models do not show the loss of motor coordination and communication skills seen in people with the syndrome.
Some researchers are manipulating MECP2 in specific cells or regions of the mouse brain to pinpoint the source of Rett syndrome traits. Deleting the gene only in inhibitory neurons, which dampen brain signaling, can produce most of the syndrome’s traits, including social deficits and motor problems. So, the signaling molecule gamma-aminobutyric acid, which is released from inhibitory neurons, is likely to be important in Rett syndrome, as it is in autism.
Mice lacking MECP2 in specific types of inhibitory neurons or in excitatory neurons have other combinations of Rett traits. Deleting genes from specific cell types or brain regions could also help researchers determine how autism features arise in the brain.
What else do researchers know about the Rett syndrome gene?
MECP2 regulates the expression of thousands of other genes. Studying its function could help scientists understand other autism genes, such as CHD8, that are ‘master regulators’ of gene expression. MECP2 also regulates the expression of a large number of unusually long genes, some of which are known to be involved in autism.
At least a dozen other genes that MECP2 controls have also been implicated in autism. These genes include FOXP1, GABRA3 and TBR1, and they may provide clues to molecular pathways involved in autism.
Could studies of Rett syndrome lead to autism treatments?
Scientists aim to treat Rett syndrome using drugs to counteract the effects of MECP2’s absence on neurons, or with gene-therapy approaches that normalize brain levels of the protein.
Manipulating MECP2 is unlikely to help people with other forms of autism. But identifying brain circuits common to both conditions could point to treatments, such as deep brain stimulation, that target the circuits.