This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
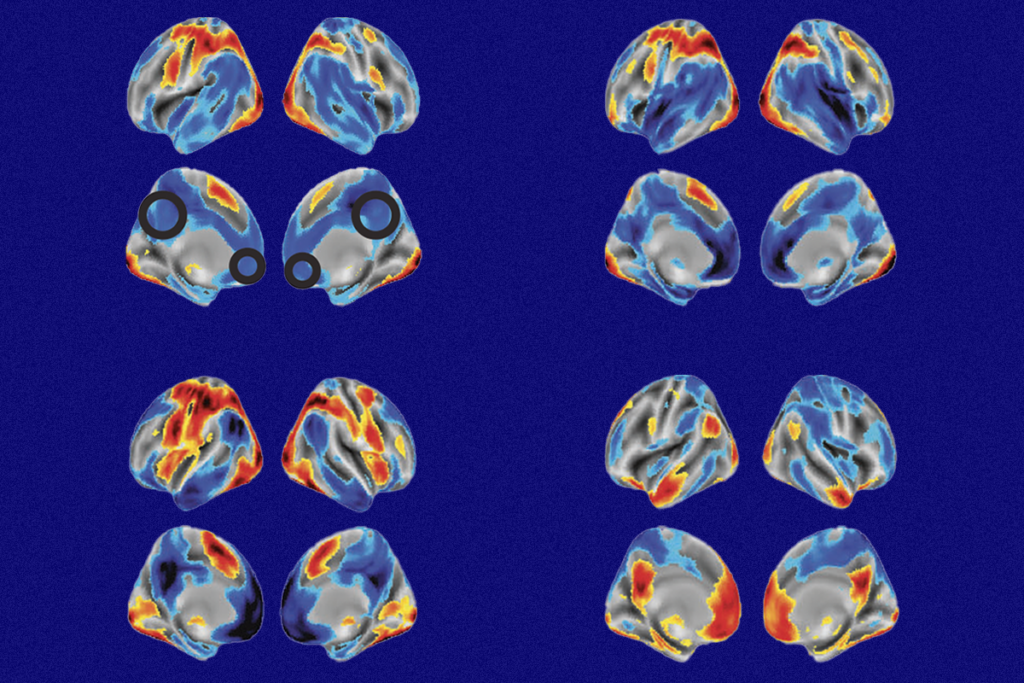
Removing the Rett syndrome gene, MeCP2, from distinct cells and brain regions reveals hidden features of the condition.
The gene mutated in Rett syndrome, a condition closely linked to autism, is called MeCP2. It regulates the expression of thousands of other genes and is expressed throughout the body, including the brain.
Removing MeCP2 from distinct cells and brain regions is one way to reveal the hidden features of the syndrome and discover new treatment targets. Several studies over the past eight years have found, for example, that the hypothalamus may be responsible for the anxiety seen in Rett.
Girls with Rett syndrome show a complex array of features, including early loss of skills, seizures, motor problems and an inability to talk. The few boys with the syndrome typically die by early childhood. A significant proportion of girls with the syndrome are also diagnosed with autism.
Mouse studies have revealed additional subtle features of Rett syndrome, such as overeating, that may be masked by more severe problems in people.
Geneticist Huda Zoghbi first identified mutations in MeCP2 as Rett’s cause in a 1999 study1. Her lab has been at the forefront of work eliminating the gene from distinct brain regions.
We asked Zoghbi, professor of molecular and human genetics at Baylor College of Medicine in Houston, Texas, about the power of this approach to study Rett syndrome.
Spectrum: What did you learn by removing MeCP2 from select brain regions?
Huda Zoghbi: We first removed MeCP2 from the mouse hypothalamus, a brain region that regulates the response to hunger and stress, among other functions. This led to overeating, aggression and anxiety.
These features are difficult to detect in some people with Rett syndrome. For instance, someone who is severely affected may not overeat because she can’t walk to the fridge or chew food efficiently.
After we published this mouse study, families started to contact me about their children who have much milder features than we’d seen before, probably due to less damaging mutations in MeCP2. These children ate too much and showed aggression and social difficulties reminiscent of what we saw in our mice. This suggested to me that the neural circuits involved in feeding and social behavior are among the most vulnerable to MeCP2 dysfunction, as they manifest in these mild mutations.
It’s amazing how much I learned from that one experiment. It taught me that we could gradually uncover subtle features of Rett syndrome by removing MeCP2 from specific brain regions and circuits.
S: What happens when you remove MeCP2 from specific cell types?
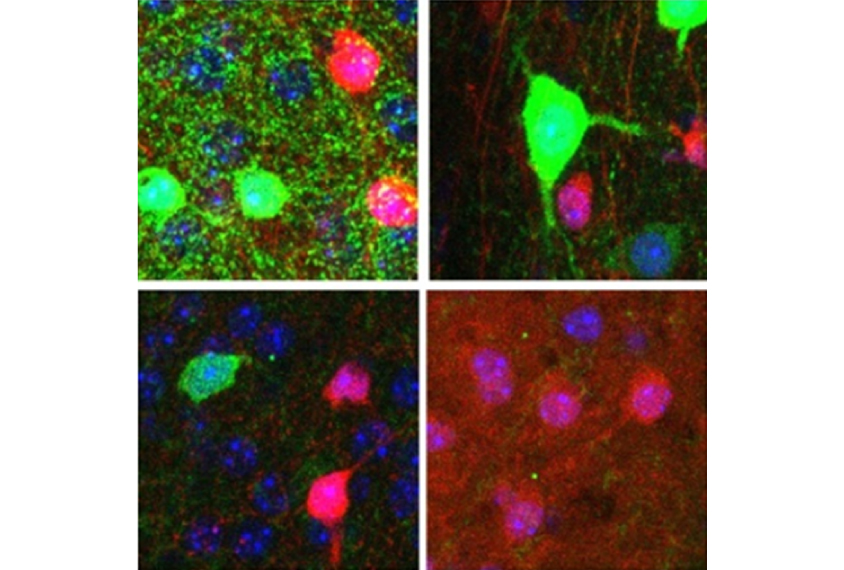
HZ: In a 2010 study, we removed MeCP2 specifically from a family of neurons that dampen brain signaling. Truthfully, I was surprised by the results. I always knew these so-called inhibitory neurons were important, but I did not expect the mice to be as impaired as they were. These male mice showed most of the behavioral features of Rett syndrome, such as social deficits and motor problems, and died prematurely.
In a 2015 study, we removed MeCP2 from each of two types of inhibitory neurons, which express markers called parvalbumin and somatostatin. We found that mice lacking MeCP2 in parvalbumin neurons have problems such as social deficits and motor impairments. Mice lacking MeCP2 in somatostatin neurons tend to have seizures.
In a study published last year, we showed that mice that lack MeCP2 in excitatory neurons — which boost brain activity — are anxious. That’s something we never saw with the inhibitory mutants.
Studies like these tie clinical features to vulnerabilities in certain types of neurons. They also hint at how neurons work together in a network. If you find the same feature when removing MeCP2 from different types of neurons, it suggests those neurons are part of the same circuit.
These studies also teach us something about neuropsychiatric conditions more generally. People with these conditions tend to have problems with signaling between neurons, rather than a wholesale loss of neurons. Image any brain you want — whether from a person with autism, intellectual disability, bipolar disorder or schizophrenia. All of these people have the right hardware. It’s the software — how the neurons communicate — that’s altered.
S: Do you see different responses in male and female mice?
HZ: MeCP2 is on the X chromosome. So males with a MeCP2 mutation have no functional MeCP2 protein. Females, on the other hand, randomly inactivate one X chromosome throughout their bodies. They will have some cells with typical levels of MeCP2 protein, and some with no functional protein.
Female mice have pretty much all the same features as males, but to a milder degree, and they have a normal life span. But there are differences in their brain networks. The network behaves differently when there is a mixture of typical and mutant cells than it does when all the cells are mutant.
What’s most fascinating to me is that restoring MeCP2 function to certain neurons in mutant mice has a different effect in males than in females.
In mutant female mice, restoring MeCP2 only in neurons that boost brain signaling alleviates more symptoms than restoring it only in inhibitory neurons. In males, the opposite is true: Restoring MeCP2 function to inhibitory neurons rescues more features. This suggests that the best treatment for boys who totally lack the gene might not be the best one for girls.
S: Does your work hint at treatments for Rett syndrome?
HZ: That’s what I’m scratching my head about every day. We know, for example, that neurons that release the chemical messenger serotonin are implicated in Rett syndrome. And we know we can boost the serotonin system using drugs developed for depression. But from what I’ve learned, I don’t think you can boost one system in isolation. You have to think about all the components.
The questions become, do we want to boost every chemical messenger, and to what degree? And is it safe to boost them constantly, or can we do it intermittently?
We would have to make a cocktail of different drugs and test for benefits. The trial would be a nightmare to do. We would have to test the combination, and then drop different drugs to see which ones are necessary. But at least it’s something to keep in the back of our minds.
Another idea is to stimulate specific neural networks to improve their function. This is why we began investigating deep brain stimulation. We see nice results in female MeCP2 mutant mice when we stimulate neurons in the hippocampus.
The best option, I think, is to find a way to restore MeCP2 expression in the brain. If we can somehow bring the protein back at the desired dose, where it’s not massively overexpressed, that would be the ideal treatment for Rett syndrome.