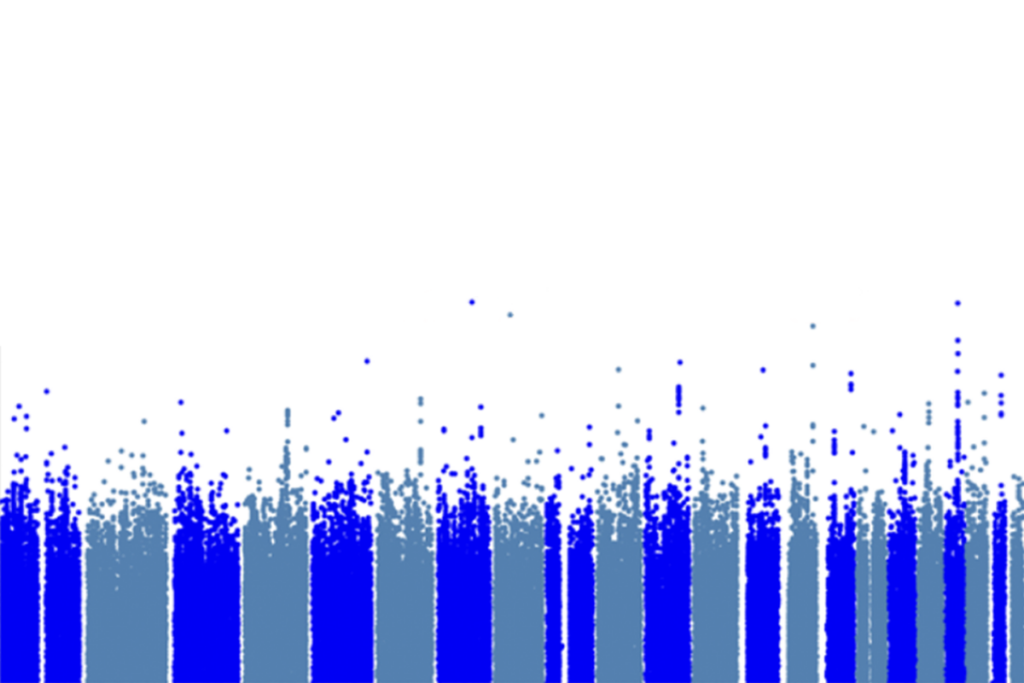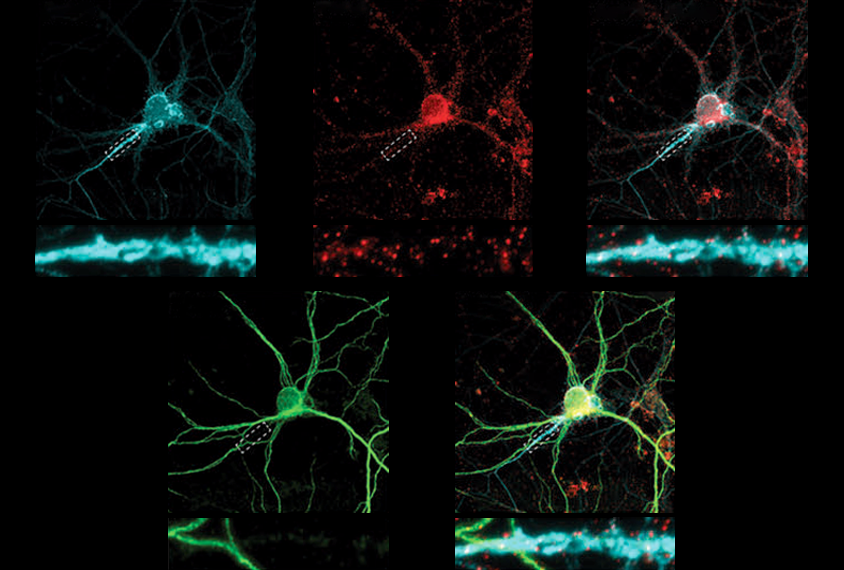
Faulty interactions between OTUD7A and other proteins, including autism-linked ankyrins, may explain traits associated with microdeletion, a type of copy number variation (CNV), in the 15q13.3 chromosomal region, according to a study published last month in Molecular Psychiatry. The altered protein network appeared to hinder neurons from clearing old proteins and deform the axon initial segment, which regulates excitability.
“Within the neuroscience field, we felt like there’s room to kind of take the approach of using cell type–specific proteomics to try to understand disease pathophysiology for this CNV,” says lead investigator Karun Singh, senior scientist at the Krembil Research Institute in Toronto, Canada. “We were able to reveal a number of things that really weren’t known about this CNV specifically at all.”
“It’s a very good study, very thorough,” says Christian Schaaf, professor in human genetics at the University Hospital Heidelberg in Germany who was not involved in the work but was recently awarded a grant in collaboration with Singh and others to investigate CNVs at 15q13.3.
“It kind of solidifies the idea that OTUD7A plays a role in the pathomechanisms of 15q13 microdeletion syndrome,” Schaaf says. It “also provides further insight [that’s] interesting to see, like the interaction with [ankyrin]-3, and that will be important to study the pathomechanisms down the line.”
The team’s experiments, conducted in mice and cultured neurons derived from human stem cells, did not, however, convincingly show that OTUD7A and ankyrin-3 colocalize in neurons — a necessary prerequisite to endogenous interactions, says Matthew Rasband, professor of neuroscience at Baylor College of Medicine in Houston, Texas. “They got lots of interesting interactors, but then the devil is in the validation.”
M
icrodeletions in 15q13.3 are associated with a variety of traits, including intellectual disability, epilepsy and autism. But how the deletions contribute to such outcomes has remained a bit of a mystery.Researchers initially suspected a gene called CHRNA7 was the primary culprit, but a pair of 2018 studies pointed to a new driver gene: OTUD7A.
In the new work, Singh and his colleagues tagged any proteins that come close to OTUD7A protein in mouse and human neurons using a molecule called biotin and then identified them via mass spectrometry. Notably, several of the identified proteins localize to the axon initial segment, and some, including multiple ankyrins, are linked with autism and/or epilepsy.
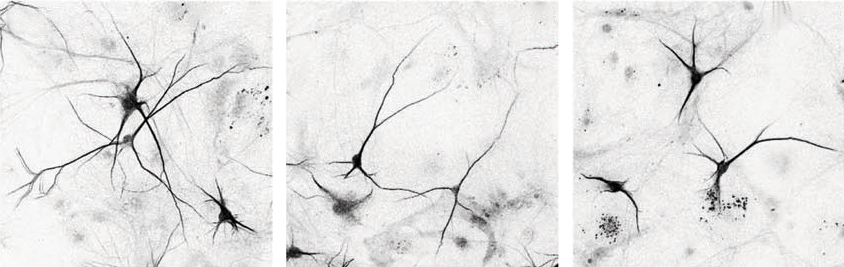
The team repeated the proteomics experiments using neurons from mice and three unrelated people with 15q13.3 microdeletions as well as one person with a mutated copy of OTUD7A. Some of the identified proteins, including ankyrin-2 and ankyrin-3, were lost from the network — in support of the idea that OTUD7A interacts with them, Singh says. These mutation-carrying neurons also showed structural alterations, which resolved in 15q13.3 microdeletion neurons that were treated with OTUD7A.
And in a new mouse model, which the team engineered in order to target OTUD7A using fluorescently tagged antibodies, the protein also overlapped with fluorescently tagged ankyrin-3, including in the axon initial segment where ankyrin-3 is known to accumulate.
“We are pretty confident there is an interaction (or close proximity) based on the approaches we have used,” Singh told Spectrum in an email. “We never claim it [is] a direct interaction/association,” he adds, but independent findings support the idea that it might be: A recent preprint that used endogenous proximity labeling of ankyrin-3 in mouse brains did find a direct interaction with OTUD7A.
“The proximity biotinylation approach is a very, very powerful approach,” Rasband says. “And for me, it was exciting to see that they found a potential proximity proteome” that partially overlaps with one he and his colleagues published in 2020. But, he says, it doesn’t prove the proteins were interacting.
The analysis of neurons derived from someone with a OTUD7A variant “provides further evidence that also point mutations or single nucleotide variants in OTUD7A can be disease causing or disease associated,” says Schaaf, co-chair of ClinGen’s Intellectual Disability and Autism Gene Curation Expert Panel — something that is not yet recognized in Online Mendelian Inheritance in Man (OMIM), a catalog of human genes and genetic disorders and traits.
“I would expect that soon enough it will be listed in OMIM,” he says, although more people must be identified to bolster the evidence.
P
revious studies suggest that OTUD7A functions as a deubiquitinase, an enzyme that removes ubiquitin tags that cells use to identify old proteins for degradation. This is a key part of the cell’s protein homeostasis pathway, which has previously been linked to autism. Looking at OTUD7A’s potential role in this pathway, Singh and his colleagues found that some of the proteins identified in OTUD7A’s proximity proteome seemed to accumulate ubiquitin in the neurons derived from people with OTUD7A mutations or 15q13.3 microdeletions and, as a result, stuck around longer.“It contributes to the growing evidence that that protein homeostasis is a dysfunctional pathway” in autism and other neurodevelopmental conditions, Singh says. “[S]ince this was done in patient-derived neurons, we think the data gives a better representation of what is happening in the human 15q13.3 microdeletion condition.”
The deubiquitination mechanism is “still something that needs to be investigated further,” Schaaf says, perhaps through broader assays to look at ubiquitination profiles across the proteome in response to a knockout or knockdown of OTUD7A.
“I don’t see a big change in the ubiquitination at all,” Rasband adds. “It’s just hard for me as a card-carrying molecular neurobiologist who does lots of these kinds of studies to see the significant differences.”
Future study may consider other genes in the 15q13.3 segment, Singh says. “It was a bit of a biased approach because we only studied one gene in the CNV.… We don’t know the contribution of other genes yet.”
His group also plans to study these mechanisms in three dimensions, using patient-derived brain organoids, to look at not just neurons but other cell types, such as glia, and to study early timepoints in neuronal development, he says.