Tiny balls of cultured human brain cells reveal how genetic mutations linked to Pitt-Hopkins syndrome, a profound form of autism, disrupt brain development, according to a new study. These balls of cells — called brain organoids — also reveal how gene therapies might help lessen the mutations’ effects.
“The data offers hope that a future clinical application with gene therapy might actually benefit certain autism spectrum disorder patients,” says co-lead investigator Alysson Muotri, professor of pediatrics and of cellular and molecular medicine, and director of the stem cell program, at the University of California, San Diego. “Our research is focused on profound autism spectrum disorder subtypes — individuals that really need help and medical support to achieve their full potential.”
Pitt-Hopkins syndrome is caused by de novo mutations in TCF4, a gene that controls when other genes are turned on or off in the embryo and is essential for brain development. Children with Pitt-Hopkins syndrome have severe cognitive and motor disabilities and typically are nonverbal.
“There is a rough estimate that Pitt-Hopkins syndrome affects 1 in every 30,000 individuals, but this is an underestimate, because genetic diagnostic testing is not available in most countries,” says co-lead investigator Fabio Papes, associate professor of genetics, evolution, microbiology and immunology at the University of Campinas in São Paulo, Brazil.
Papes has a relative with Pitt-Hopkins syndrome, which spurred him to study how TCF4 mutations impair brain development. He and Muotri turned to organoids because mouse models of the condition are limited: The animals display some Pitt-Hopkins syndrome-like traits, such as impairments in social interactions and memory, but fail to mimic the severe traits seen in people.
The new results highlight “once more, the need for human models in order to fully understand the genetics of neurodevelopmental disorders,” says Jürgen Knoblich, scientific director of the Institute of Molecular Biotechnology at the Austrian Academy of Sciences in Vienna, Austria, who did not take part in this research. It “represents nice and useful progress for the field of neurodevelopmental disorders.”
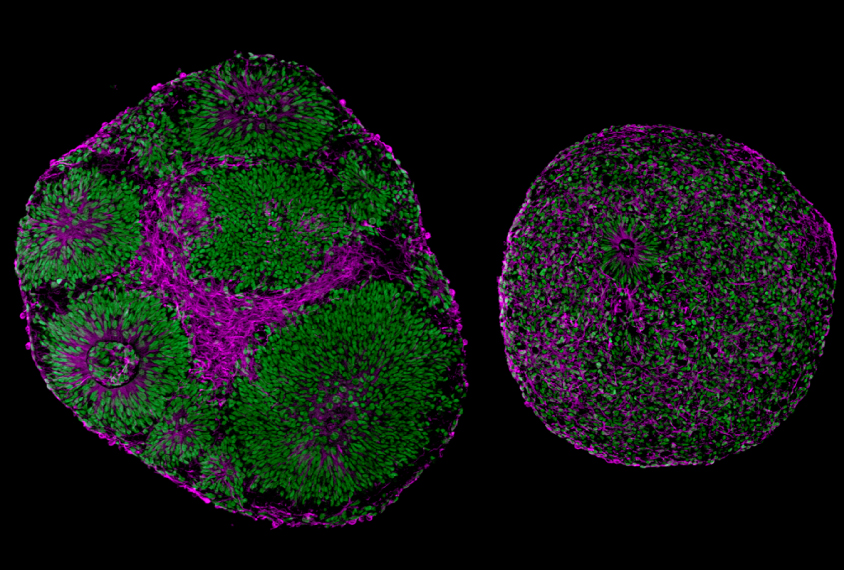
M
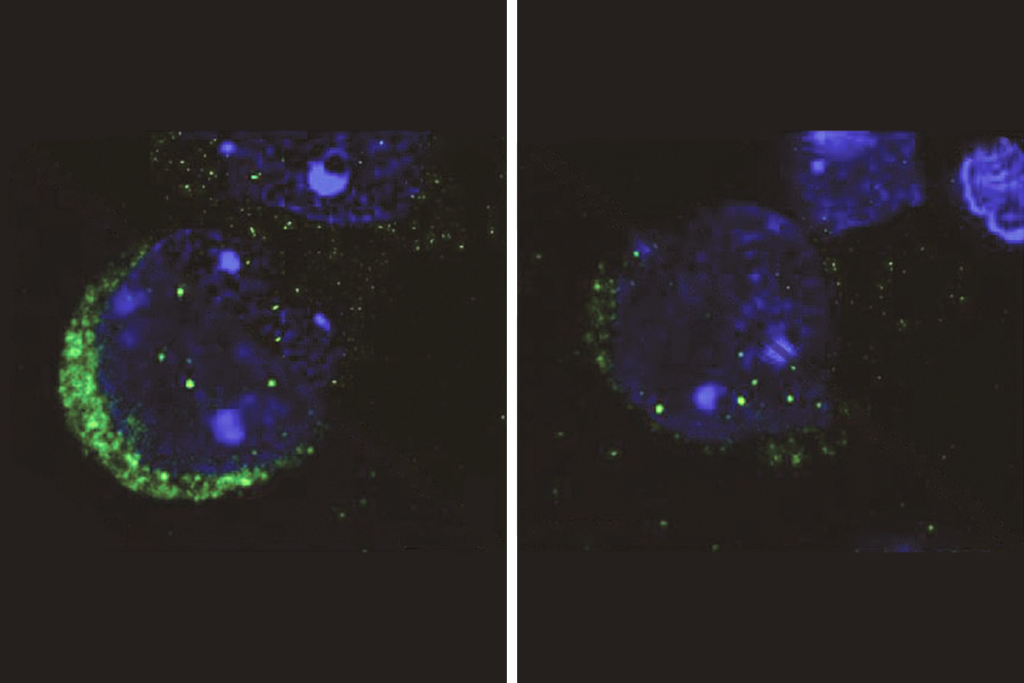
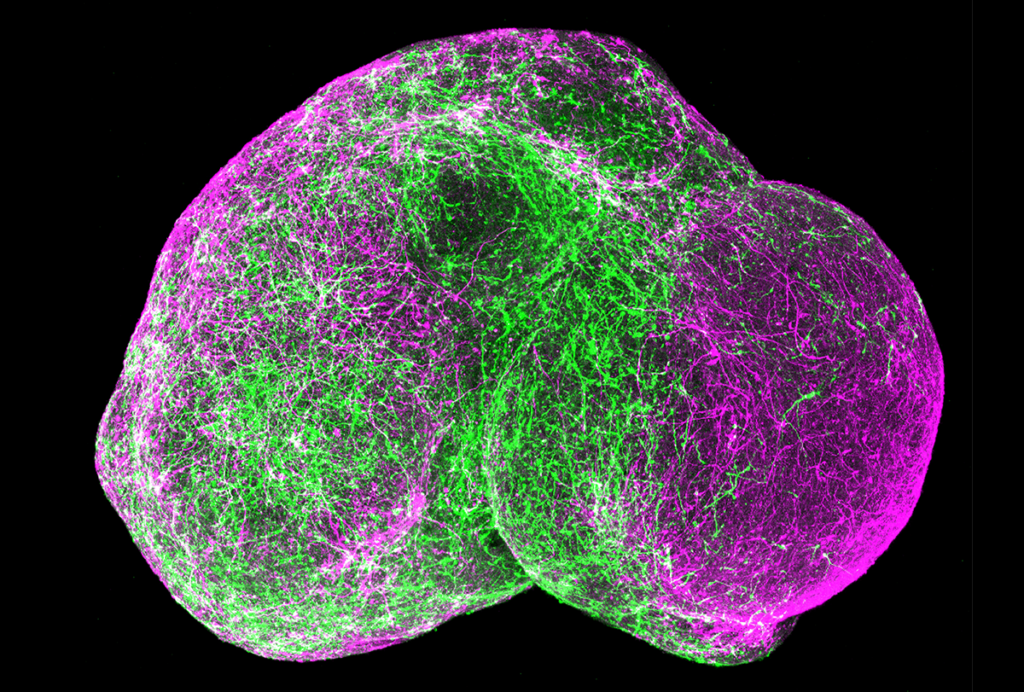
uotri, Papes and their colleagues generated brain organoids using skin cells from five children with Pitt-Hopkins syndrome and five controls.After four weeks, organoids with TCF4 mutations were significantly smaller than those without, and many of their cells had not developed into neurons from precursor cells called neural progenitors.
The neural progenitors exhibited arrested development, likely due to a disruption in a cell signaling pathway called WNT and then reduced expression of a gene called SOX, the scientists found. Compared with controls, the progenitors yielded fewer cortical neurons, which power thinking, memory, speech and other cognitive abilities. And the neurons they did produce showed impaired electrical activity and tended to cluster together instead of self-organizing into neural circuits. The findings appeared in Nature Communications in May.
It is surprising to see such significant effects on neural progenitor cells, because the obvious traits of Pitt-Hopkins syndrome, such as microcephaly, or a small head, usually develop after birth, says Ben Philpot, professor of cell biology and physiology at the University of North Carolina at Chapel Hill, who did not work on this study. “This suggests that their organoid model was highly sensitive and has great value for screening potential therapeutics,” he says.
The scientists also tested two different virus-based gene therapies on the organoids. One encodes molecules that boost levels of TCF4 protein, whereas the other delivers a functional copy of the gene to the cells. Both techniques resulted in organoids that resembled the controls.
“We could rescue molecular, cellular and neural network alterations by just restoring the expression of the corrected gene,” Muotri says.
I
n an unrelated study published in eLife in May, Philpot and his colleagues tested “a very similar gene therapy strategy in mouse models,” Papes says. “Both papers complement each other very nicely, providing very solid evidence, in patient-derived human cells and in animals, that gene therapy is possible for this form of profound autism.”Muotri, Papes and their colleagues have partnered with a pharmaceutical company specializing in gene therapy to use these new findings to design a clinical trial. “The clinical potential is huge,” Muotri says. “We will start with TCF4, but our strategy allows us to test other genes implicated in autism spectrum disorder.”
The scientists caution that these findings may not translate for people with Pitt-Hopkins syndrome. “A brain organoid is not a ‘mini-brain,’” Muotri says. “These are really small structures when compared to the human brain. Also, they are mimicking developmental stages rather than a more mature human brain.”
In addition, children enrolled in a clinical trial would receive the gene therapies a few years after the prenatal stage of brain development in which the therapies were tested. “Our gene therapy approach may not correct all characteristics, especially those changes that cannot be reverted once the cells and brain tissue have already been altered,” Papes cautions.
Before clinical trials can ever happen, “we need to be absolutely convinced that our methods will cause no harm and will be effective,” Muotri says. “Thus, we have a lot of work to do in the following years.”