This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
The autism-linked gene SHANK3 is known for its role at neuronal junctions, but it has another function that could serve as a drug target.
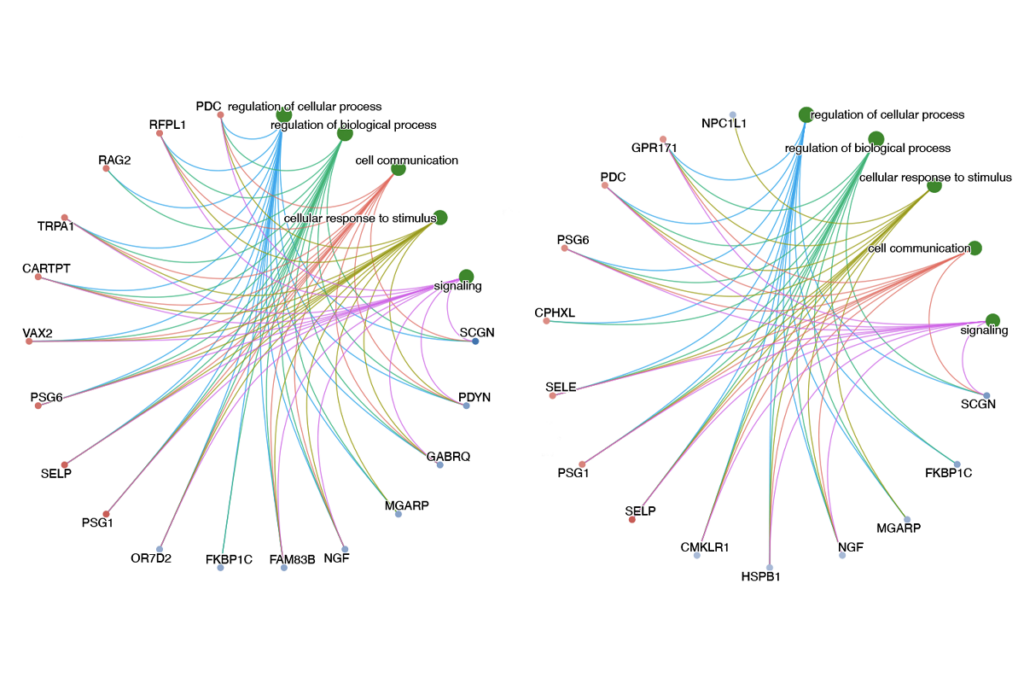
The autism-linked gene SHANK3 has a previously undiscovered role in neurons that hints at a new drug target, suggest findings published 10 March in Science1. Mutations in the gene occur in up to 2 percent of people with autism and intellectual disability.
SHANK3 is known for structurally supporting synapses, the communication hubs between neurons. Its role at synapses has contributed to theories of autism as a ‘synaptopathy’ — a condition caused by defects in neuron signaling.
The new study suggests that SHANK3’s function extends beyond these tiny junctions. It shows that neurons missing the gene have faulty ion channels throughout their outer membranes. These channels play a critical role in neuronal signaling, as they regulate overall neuron excitability.
“You can’t just assume that a gene that encodes a protein which is synaptic will necessarily cause a synaptic deficit, and only a synaptic deficit,” says lead investigator Thomas Südhof, professor of molecular and cellular physiology at Stanford University in California.
Südhof’s team found that SHANK3 binds to a common class of ion channels, known as HCN channels, that open in response to electrical signals, allowing charged molecules to flow in and out of the cell. This process helps to set the neuron’s ability to conduct electrical signals. Neurons missing SHANK3 have fewer of these channels and tend to be unusually excitable.
The study may reinvigorate efforts to develop drugs for conditions tied to SHANK3 mutations, says Thomas Bourgeron, professor of genetics at the Institut Pasteur in Paris, who was not involved in the study. “It might be easier to target the HCN channels than to target [SHANK3] itself.”
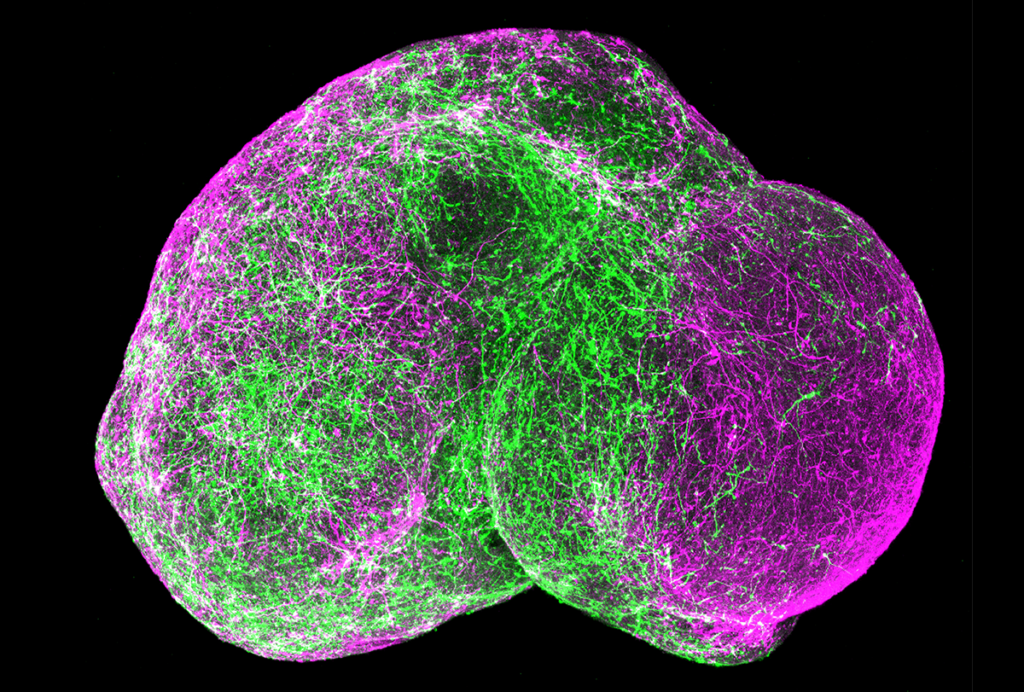
The first hints that SHANK3 has a role beyond synapses came from a 2013 study in which researchers generated neurons from the skin cells of people with Phelan-McDermid syndrome, who lack a portion of chromosome 22 where the SHANK3 gene resides2.
These neurons have fewer synapses and transmit fewer and weaker excitatory electrical messages than do those from controls, that study found. Introducing a copy of SHANK3 normalizes these problems, confirming that SHANK3 is required for synapse function.
But the study also found that the mutant neurons tend to be more excitable than control neurons. The researchers assumed that this hyperexcitability somehow stemmed from the loss of synapses.
“We didn’t know how these two were connected,” says Alex Shcheglovitov, assistant professor of neurobiology and anatomy at the University of Utah in Salt Lake City, who was involved in the 2013 work. One theory was that to compensate for the decrease in excitatory signaling, the neurons became more excitable.
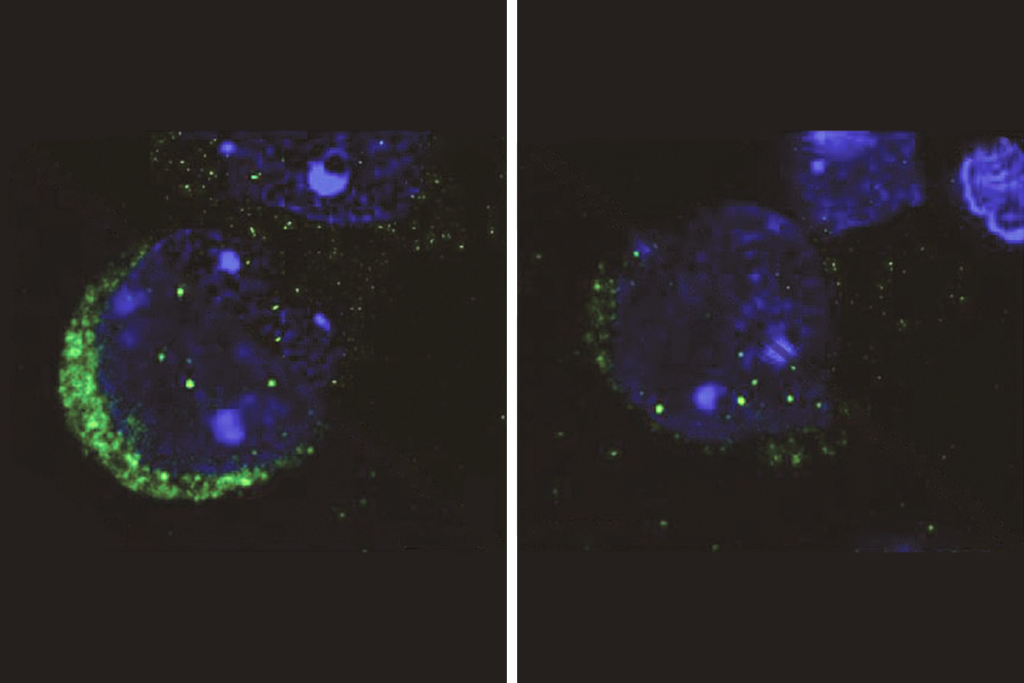
In the new study, Südhof and his colleagues created human embryonic stem cells with either one or two faulty copies of SHANK3. They then coaxed the mutant stem cells into becoming neurons by engineering them to express a particular gene.
They found that the SHANK3-deprived neurons sprout fewer and shorter dendrites, neuronal branches that receive signals at synapses, compared with controls. Like the Phelan-McDermid cells, these neurons also transmit about half the excitatory signals as controls do and, at the same time, show as much as a 33 percent increase in their excitability.
Südhof’s team then measured the flow of current through various types of ion channels known to determine a neuron’s excitability. The only change they detected was a lower current through HCN channels in the SHANK3 mutants than in controls.
SHANK3 binds to these channels in cultured kidney cells, and chemically blocking the channels in control neurons mimics the effects of SHANK3 loss, the researchers found. Together, the findings suggest that the loss of SHANK3 impairs the function of HCN channels.
It is still unclear whether restoring the function of the channels would return the mutant cells’ excitability to normal, however.
“That would actually tell you that this is the key node in this pathway and that, perhaps if you made an activator to this channel, you could rescue the phenotypes,” says Ricardo Dolmetsch, head of neuroscience at Novartis Institutes for Biomedical Research in Cambridge, Massachusetts. Dolmetsch led the 2013 study but was not involved in the new work.
It is also uncertain whether the faulty channels have anything to do with symptoms seen in people who lack SHANK3. Intriguingly, impairments in HCN channels can lead to difficulty detecting pain, a feature found in some people with Phelan-McDermid syndrome.
Having hyperexcitable neurons could also raise the risk of seizures, a symptom of Phelan-McDermid syndrome, says Craig Powell, associate professor of neurology and neurotherapeutics at the University of Texas Southwestern in Dallas, who was not involved in the study.
The next step is to find out whether the study’s findings hold true for neurons in living mice missing SHANK3. “Although these are human neurons, they are neurons in culture,” Powell says. “Neurons in culture often behave differently — especially in their electrical properties — than they do in brain tissue.”