This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
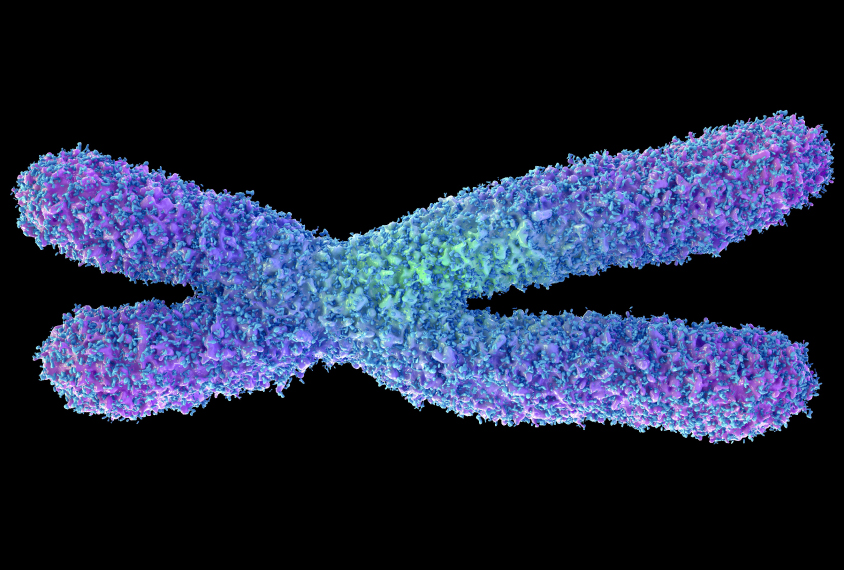
Manipulating pathways that help silence the X chromosome may help treat Rett syndrome, a condition closely related to autism.
Manipulating pathways that help silence a copy of the X chromosome may help treat Rett syndrome, a condition closely related to autism, according to a new study1. The study identified 30 key genes in these pathways.
The results offer a proof of concept. But they also represent the first step to developing new treatments for the condition, says lead investigator Antonio Bedalov, associate professor of medicine at the University of Washington in Seattle.
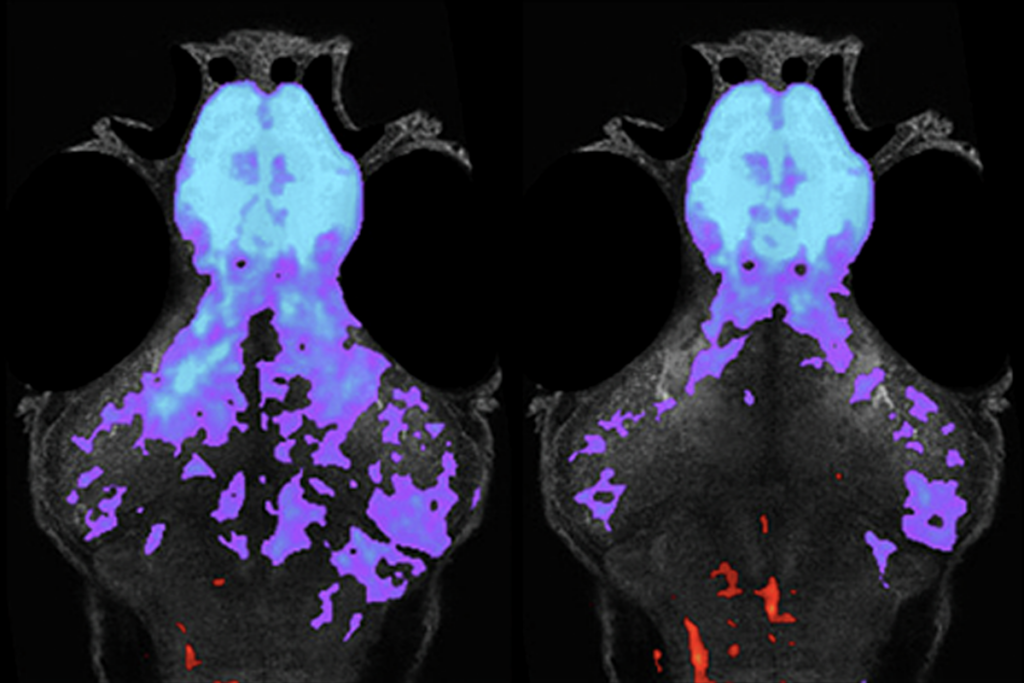
Rett syndrome stems from mutations in MeCP2, a gene on the X chromosome. Girls with Rett syndrome have only one mutant copy of the gene. But due to a process called random X inactivation, the second — and intact — copy of MeCP2 is silenced in some of their cells.
In the new study, published 14 February in the Proceedings of the National Academy of Sciences, researchers were able to unmute this silent copy. But it is unclear exactly how much MeCP2 is enough. “We don’t really know how much to reactivate to confer therapeutic benefit,” Bedalov says.
The study illustrated the reactivation concept in mouse cells. It is not yet clear whether the same approach would work in people.
Still, “the proof-of-principle that MeCP2 can be unsilenced is exciting and promising,” says Benjamin Philpot, associate director of the Neuroscience Center at the University of North Carolina at Chapel Hill, who was not involved in the work.
Researchers have for years batted around the concept of unmuting a silent, typical gene copy for Rett, as well as for other autism-related conditions, such as Angelman syndrome.
A 2014 study revealed that in the case of the X chromosome, 15 genes are required for silencing the chromosome. The new work highlights four of the same genes and some of the same pathways the previous study identified.
Bedalov and his team made mice in which MeCP2 is fused to a gene that confers resistance to the antibiotic hygromycin. They isolated cells in which this fusion is silent. They then exposed those cells to thousands of small pieces of RNA, each of which inactivates one of 25,000 genes.
Only cells that express the resistance gene survive in the presence of hygromycin. The researchers identified the piece of introduced RNA in each surviving cell, leading them to pinpoint 30 genes. Six of the genes are involved in a pathway needed for forming bones; one of these genes, ACVR1, also turned up in the 2014 study.
Another gene in the same family, RNF12, is known to play a role in X chromosome silencing during early development. The researchers showed that RNF12 retains its silencing role in mature cells.
“It’s nice that [Bedalov] found factors and pathways that we had identified, but then found additional ones as well,” says Michael Green, professor of molecular medicine at the University of Massachusetts Medical School, who led the 2014 study.
The new study points to RNF12 as a possible therapeutic target for Rett syndrome. But some experts caution that disrupting RNF12 could have unintended consequences: Mutations in the gene are associated with intellectual disability, notes Ype Elgersma, professor of neuroscience at Erasmus University in Rotterdam, the Netherlands, who was not involved in the study.
Unmuting the silent X chromosome also affects genes other than MeCP2, the researchers found.
When the researchers can show that they are able to specifically unmute only MeCP2, “I will become even more excited,” says Gail Mandel, professor of biochemistry and molecular biology at Oregon Health and Science University in Portland, who was not involved in the work.
Bedalov and his team plan to make female mice that have one mutant copy and one typical, silenced copy — and then test their approach in those mice.