Some mutations in SCN2A, a gene reliably linked to autism, change social behaviors in mice by dampening the electrical activity of their neurons, according to a new study.
SCN2A encodes a sodium channel that helps neurons send electrical signals. So-called ‘gain-of-function’ mutations make the channel hyperactive and can lead to epilepsy, whereas ‘loss-of-function’ mutations diminish its activity and are typically associated with autism.
The mice in the new study carry the latter type and, as a result, have fewer functioning sodium channels than usual. The animals also react to unfamiliar mice in an atypical way, mirroring social behaviors seen in autistic people with similar SCN2A mutations.
“We’re in the position of really connecting a single mutation, or at least a defect in the channel, to the behavior,” says lead investigator Geoffrey Pitt, professor of medicine at Weill Cornell Medicine in New York. “The message that our paper shows is that loss-of-function mutations and decreased sodium current can lead to behaviors.”
This study confirms previous work showing that autism-linked mutations in SCN2A dampen channel activity in neurons, and further connects the loss-of-function mutations to clear changes in behavior, says Kevin Bender, associate professor of neurology at the University of California San Francisco, who was not involved in the work. “The behavioral results were actually some of the most robust that I’ve seen in this field to date.”
Low sodium:
Pitt and his team used CRISPR to make mice carrying an SCN2A mutation that effectively eliminates a portion of the sodium channel protein.
This mutation has not been observed in people, Bender says, but it mimics the truncated proteins found in most people with loss-of-function mutations in SCN2A.
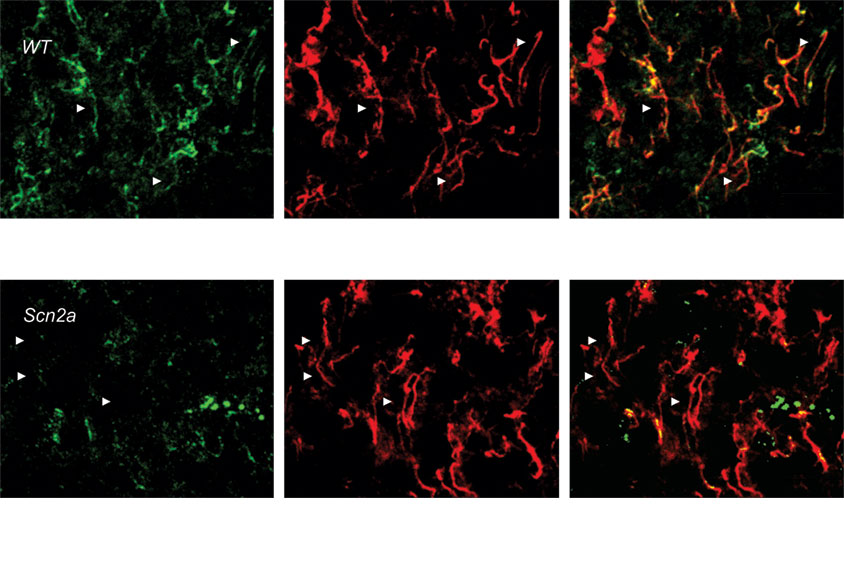
Mice with the mutation in both copies of the gene die shortly after birth and have almost none of the sodium channel in their cerebral cortex, Pitt’s team found. So they focused on mice with one mutated copy of SCN2A, which have a normal lifespan and less than half the usual amount of the sodium channel.
Pyramidal neurons isolated from the animals’ forebrains were less excitable than those from wildtype mice. Pitt and his colleagues also used fiber photometry to monitor the activity of these neurons in the medial prefrontal cortex as the mice moved around in a plus-sign-shaped ‘maze’ with two arms that are enclosed and two that are not.
Typical mice tend to spend less time in the open arms, where they become anxious. They also show a marked increase in neuronal activity while visiting the open arms. By contrast, the SCN2A mice extensively explored the open arms and showed almost no change in neuronal activity when moving between the open and enclosed arms.
“The fact that these cells have no difference in their activity in vivo is quite remarkable,” Bender says. “That’s something that would be really interesting to follow up on.”
In a test of the animals’ social behaviors, the SCN2A mice were just as interested in an unfamiliar mouse as wildtype mice were, but they remained so for far longer. These behaviors line up roughly with clinical data from four autistic people with loss-of-function SCN2A mutations, who tend to be overly friendly with strangers, the researchers say.
In further analyses, brain slices from the model mice’s medial prefrontal cortex and basolateral amygdala — areas associated with cognitive and emotional behaviors — showed less frequent spontaneous excitatory postsynaptic currents than did brain slices from controls. That finding suggests that the decrease in sodium channels reduced neuronal firing, leading to impaired neuronal signaling.
“It’s heartening to see potential consistencies between the clinical data and some aspects of our mouse model,” Pitt says. His team’s next step is to evaluate mice with a loss-of-function SCN2A mutation found in autistic people, he says.