Brain cells from mice that model fragile X syndrome — the most common inherited cause of autism — show excessive protein breakdown, a new study has found. And normalizing that process reverses other molecular alterations — suggesting new avenues for treatment strategies.
The findings add to mounting evidence that challenges a long-standing theory about the condition — namely that its traits stem primarily from protein overproduction. But the results are not surprising because protein production and degradation are linked, and defects in one can lead to compensatory changes in the other, says Gary Bassell, professor of cell biology at Emory University in Atlanta, Georgia, who was not involved in the study.
“In a lot of cases in autism, there’s an effort by neurons in a circuit to try to compensate for a molecular lesion or deficiency in certain genes,” Bassell says. “In many cases, the compensation works, but in some cases, it leads to altered synaptic plasticity.”
Fragile X syndrome arises from mutations that prevent the production of a protein called FMRP that curbs the synthesis of other proteins.
Mice that lack FMRP overproduce proteins, particularly at synapses. Having too many synaptic proteins is thought to disrupt neuronal function. But cells derived from people with fragile X syndrome or mice that model the condition do not show dramatic increases in total protein levels.
“That was always a puzzle,” says Emily Osterweil, professor of molecular neuroscience at the University of Edinburgh in Scotland, who led the new work.
Osterweil and her team expected that excessive protein production would drive a compensatory rise in protein breakdown by the proteasome — a molecular machine that chops up proteins carrying a specific chemical tag.
“What we didn’t expect was that [increased protein degradation] was going to cause its own problems,” Osterweil says.
I
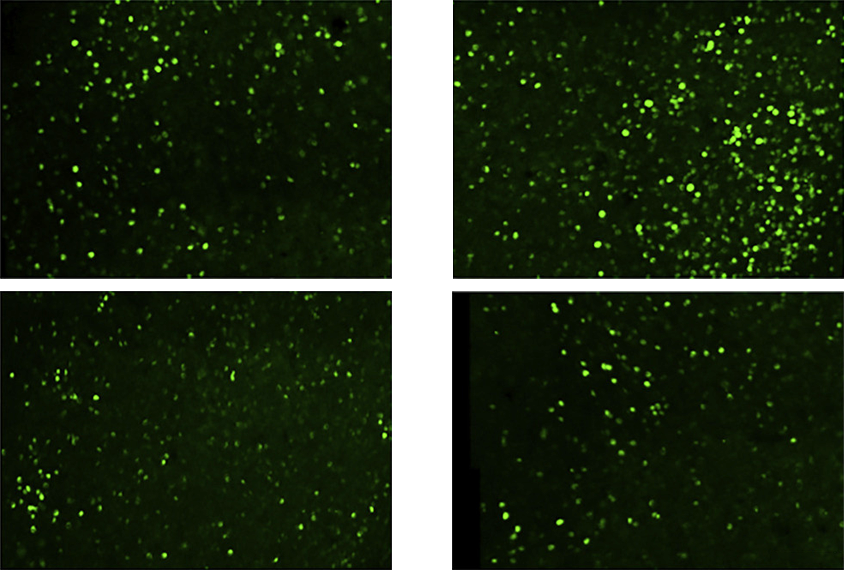
n mice lacking FMRP, the proteasome excessively degraded proteins in the hippocampus, a brain region central to learning and memory, including two subunits of a protein important for multiple synaptic receptors.“It’s possible that increasing the turnover of those proteins would alter the function of many receptors,” Osterweil says. “That’s one case where this increased protein breakdown would be a problem.”
Previous studies have shown that inhibiting the proteasome reduces new protein synthesis in neurons. Indeed, lowering proteasome activity with bortezomib, a drug used for cancer treatment, reduced excessive protein production in the hippocampus of the fragile X mice, Osterweil’s team found.
In the same mouse model, proteasome activity was also increased in the inferior colliculus, a brain area that processes sound-related information and contributes to ‘audiogenic’ seizures the animals experience in response to high-frequency sounds.
Bortezomib similarly reduced the hyperactivity of neurons in this brain area and prevented the seizures, the researchers reported last month in Neuron.
O
sterweil’s results suggest that unbalanced protein turnover may cause neurons to malfunction in fragile X syndrome, Bassell says. “A protein might have a shorter half-life — it’s being made faster, but it’s also being degraded faster, so the dynamics are altered.”But it’s unclear exactly how the turnover contributes to fragile X traits, or whether it occurs in brain regions other than the two the team focused on.
And it may be too soon to totally discard the idea that excess protein production contributes to the condition: A study posted as a preprint in December suggests that increased protein synthesis in a class of inhibitory neurons in the striatum drives repetitive behaviors in mice lacking FMRP. Blocking protein production in those neurons rescued the behaviors, the researchers found.
“Identifying a specific cell type in a specific circuit allows you to be more precise in designing therapeutics,” says study investigator Eric Klann, director of the Center for Neural Science at New York University in New York City. Drugs that dampen protein production in certain neurons may help ease some of the repetitive behaviors associated with fragile X, Klann says. “It’s ultra-precision medicine.”
B
oth findings have potential implications for the treatment of fragile X syndrome, says Baoji Xu, professor of neuroscience at the Herbert Wertheim UF Scripps Institute for Biomedical Innovation & Technology in Jupiter, Florida.Drugs that work in mice, however, may not work in people, Xu says. And it’s unclear, he adds, if inhibiting protein breakdown could normalize other traits, such as hyperactivity and social difficulties, observed in mice lacking FMRP.
Next, Osterweil plans to use models that more closely mimic the syndrome in people, such as human induced pluripotent stem cell lines, to investigate which proteins, when undergoing excessive turnover, contribute to fragile X syndrome.
“Getting at which proteins are important for maintaining synaptic excitability and plasticity — those are the next big questions,” she says.