This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
The neurons of people with Rett syndrome contain an overabundance of retrotransposons — DNA sequences that copy and insert themselves into new spots throughout the genome — during early development, according to a study published 18 November in Nature.
The neurons of people with Rett syndrome contain an overabundance of retrotransposons — DNA sequences that copy and insert themselves into new spots throughout the genome — during early development, according to a study published 18 November in Nature1.
Retrotransposons, also known as ‘jumping genes,’ make up nearly half of the mammalian genome. Long labeled as ‘junk DNA,’ these genes may have a biological function and influence disease, say researchers.
Specifically, the new results may help explain the broad range of effects in Rett syndrome, a rare and debilitating disorder on the autism spectrum caused by missing or mutated MeCP2 protein.
The disease primarily affects girls and causes a range of symptoms, such as seizures, trouble breathing, a progressive decline in social and motor abilities, and even death. The type and severity of symptoms both vary in individuals who carry the same mutation.
“There’s a lot of interest in understanding where the variability in severity of the [Rett] phenotype comes in,” says Janine LaSalle, professor of medical microbiology and immunology at the University of California, Davis, who was not involved with the work. “Are there genetic modifiers? Are there environmental modifiers? And now this study brings in a whole new aspect: Could there be retrotransposon modifiers?”
In 2005, lead investigator Alysson Muotri, then working as a postdoctoral researcher in Fred Gage’s lab at the Salk Institute for Biological Studies in La Jolla, California, discovered that retrotransposons are highly active in the brains of rodents2.
The genes are also active in human neurons, adding hundreds of extra copies of themselves to the genome during a short window in early development3. After that period, however, these genetic elements — called long interspersed nuclear element 1s, or L1 elements — tend to stay rooted in place.
Searching for molecules that control retrotransposons, Muotri’s team spotted a 2001 study showing that MeCP2 represses expression of the L1 elements in cell lines derived from human embryonic kidney cells 4.
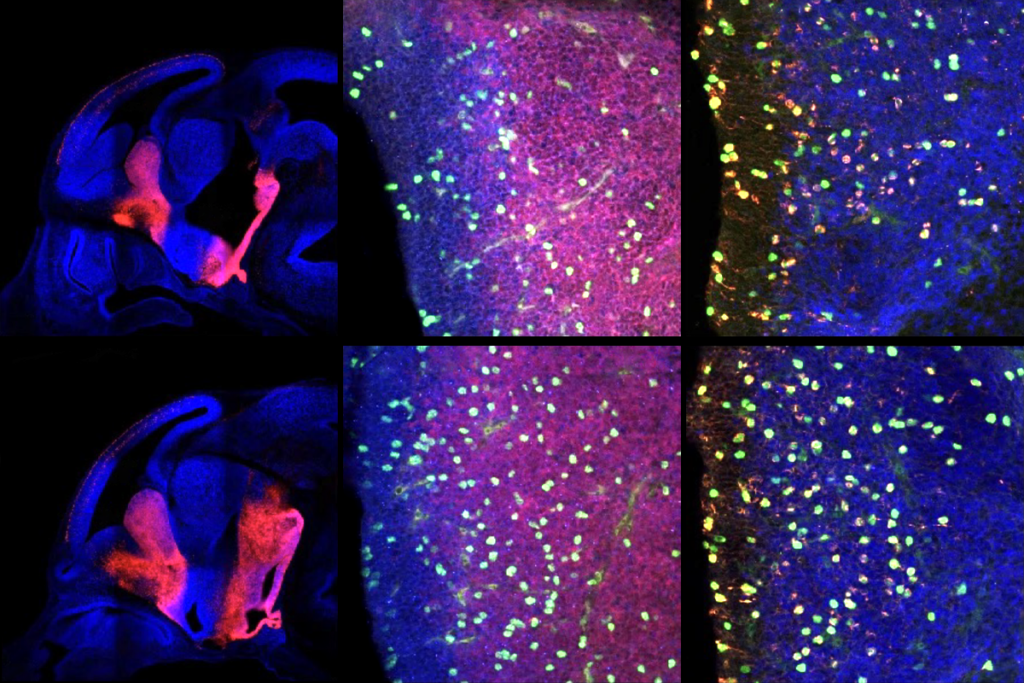
In the new study, Muotri’s group used mice lacking the MeCP2 gene whose L1 elements had been genetically engineered to express a fluorescent label. They found that L1s are roughly four times more abundant in the brains of the mutant mice compared with those of normal mice.
The results are surprising because MeCP2 is normally expressed everywhere, albeit at much higher levels in neurons, notes Muotri, now assistant professor of cellular and molecular medicine at the University of California, San Diego. “We first postulated that in the absence of MeCP2, we’re going to see increases [in L1] in all tissues of the body, but we only saw it in the nervous system,” he says.
To validate the findings in human cells, the group created a stem cell assay that transforms skin tissue into so-called ‘induced pluripotent stem cells,’ or iPS cells, which have the ability to form any type of cell5.
The researchers found that neurons derived from tissue taken from people with Rett syndrome have twice the amount of L1 compared with neurons from controls.
Analyzing postmortem brains, they also found that people with Rett syndrome have more DNA overall than do healthy controls. That suggests that L1 is more active in diseased neurons, Muotri says.
“The finding that MeCP2 mutant brains have this up-regulation of L1 elements is eye opening,” says Xinyu Zhao, associate professor of neurosciences at the University of New Mexico, who was not involved with the study. “[But] as with many things in biology, it may take some time to understand the significance of this discovery.”
Researchers still don’t know whether higher L1 levels are a cause, rather than an effect, of Rett syndrome. But the timing and location of their activity implicates them in the disorder, says LaSalle. “As much as you may argue against causality, for the phenotype of Rett it’s also hard to imagine how [L1] couldn’t be involved.”
The relationship between MeCP2 and genomic instability is part of an evolving view of MeCP2’s role in Rett. Initial studies suggested that MeCP2 binds to DNA and turns off the expression of genes. Other researchers have found that MeCP2 turns on thousands of genes, and some studies show it even participates in alternative splicing, producing slightly different forms of the protein it encodes.
In a study published in February, Adrian Bird and colleagues found evidence that MeCP2 affects the global structure of DNA by altering the way it packs into a cell6.
Muotri and colleagues are trying to understand the importance of MeCP2 in gene jumping. The first step is finding out where in the genome these copies are inserting themselves.
“We want to find out if it’s completely random, or maybe there’s a hotspot for these insertions,” Muotri says. If the insertions are found more often in neuronal or synaptic genes, for example, that would suggest they’re players in the pathology of Rett syndrome.
The group is also developing ways to inhibit the activity of retrotransposons in MeCP2-deficient mice. “We’re going to start in the mouse and if we don’t see anything in the mouse, we’re going to move to [human cells in] the iPS system,” says Muotri.