This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
Identifying the factors that protect girls from autism may help us understand and possibly treat the disorder.

The reason autism is more common in boys than in girls may be because girls are protected from the disorder in some manner. Identifying the mechanism of this protection may help us understand and treat autism.
Autism is consistently observed to be about three to four times more prevalent in boys than it is in girls. However, the reason for this difference is unclear. One simple explanation is that some autism risk factors affect only boys. Triplet repeat expansions in FMR1, the gene mutated in people with fragile X syndrome, is one such example. FMR1 is located on the X chromosome, and an expansion in the gene, which leads to intellectual disability and autism in a boy, may not produce these symptoms in a girl who has a second intact copy of the gene.
Early explorations into the risk factors for autism focused on the X chromosome for this reason. But we now know that rare and de novo, or spontaneous, genetic risk variants on the X chromosome are seen in less than 2 percent of boys with autism. Similarly, common variants on the X chromosome account for only 1.5 percent of autism risk1.
This tells us that although there are examples of male-specific genetic risk factors, their contribution to autism risk does not explain the magnitude of the sex bias in prevalence.
An alternative explanation is the female protective effect (FPE). In this model, autism risk factors affect both boys and girls, but girls are inherently protected from their effects.
We can conceptualize this model by using the analogy of height. Let’s define ‘tall’ as being in the top 1 percent of the population for height. Boys and girls can both be ‘diagnosed’ as being tall, but more boys than girls will merit this diagnosis because, on average, girls are shorter than boys. We could say that girls are protected from being tall despite being exposed to similar risks.
Based on this model we can make two predictions. First, we expect the risk factors for being tall to be shared between the two sexes. Second, we expect tall girls to have more risk factors for being tall than boys of an equivalent height.
These predictions also apply to the FPE model of autism: We expect shared risk factors for autism between the sexes and a higher overall burden of autism risk factors in affected girls than in affected boys. We can assess both of these predictions experimentally using genomic or epidemiological data.
De novo mutations are strongly associated with autism: That’s been shown for both copy number variations (CNVs) — large deletions or duplications of chromosomal regions — and for point mutations that result in a loss of function of the gene2. Identifying regions of the genome with a high burden of these de novo mutations allows us to link specific genes to autism3. Within these autism-associated genes, mutations are distributed randomly between the sexes. This evidence of shared autism risk factors matches the first expectation of the FPE model.
My colleagues and I have also seen data that match the second expectation, that girls with autism carry a higher overall burden of autism risk factors, specifically of de novo mutations. We and others have observed this for CNVs, loss of function mutations and missense mutations — point mutations that result in the substitution of one amino acid for another2,3,4,5,6.
The FPE model predicts that girls can tolerate more autism risk variants than boys. Therefore, we might expect more inherited risk variants to come from unaffected mothers than from unaffected fathers. A few groups have seen this maternal bias in rare inherited single nucleotide variants and in some, but not all, analyses of rare inherited CNVs3,7.
This lack of consistency may reflect the limited power of these analyses due to the weak association between rare inherited variants and autism, in contrast to the strong association and well-powered analyses focusing on de novo mutations.
Genomic analyses assess a small number of genetic risk factors directly, whereas epidemiological analyses assess the cumulative effects of many risk factors indirectly. Because autism is highly heritable, we expect — and observe — a higher prevalence of autism in the siblings of people with autism in epidemiological studies.
According to the FPE model, girls with autism should have a higher burden of autism risk factors than boys with autism. As a result, the prevalence of autism should be higher among the siblings of girls with autism than among the siblings of affected boys. This prediction is sometimes called the ‘Carter Effect,’ after the British geneticist who first described this phenomenon in the 1960s.
However, the majority of studies, including large population-based analyses, have not seen this sex-dependent variation in autism recurrence8. By contrast, some teams have observed results consistent with the Carter Effect and the FPE model, both for the recurrence of autism traits in the general population and in a study of autism in multiplex families, which have more than one member diagnosed with autism9,10.
Overall, the genomic evidence suggests that a female protective effect plays some role in autism’s observed sex bias. It is unclear why these data are discordant with the majority of epidemiological evidence. It is possible that de novo mutations and rare variants provide an incomplete picture of autism risk. Alternatively, the epidemiological studies may lack enough power to detect the protective effect indirectly. A third possibility is that autism risk factors interact in a more complex manner than epidemiologists assume in their studies.
One strategy for discovering the nature of the FPE is to identify factors associated with the presence of protection (for example, in unaffected girls) or the absence of protection (for example, in affected girls).
Earlier this year, my colleagues and I published the results of female-only genome-wide association studies of 1,242 girls from two collections. Our study did not identify any common variants associated with the FPE, though it was powered only to detect variants that mediate the majority of the hypothesized protection11. As we noted in the paper, our approach relied on the assumption that the degree of protection varies among girls (for example, if it is related to estrogen levels), as opposed to being the same in all girls (such as the presence of estrogen above a low threshold).
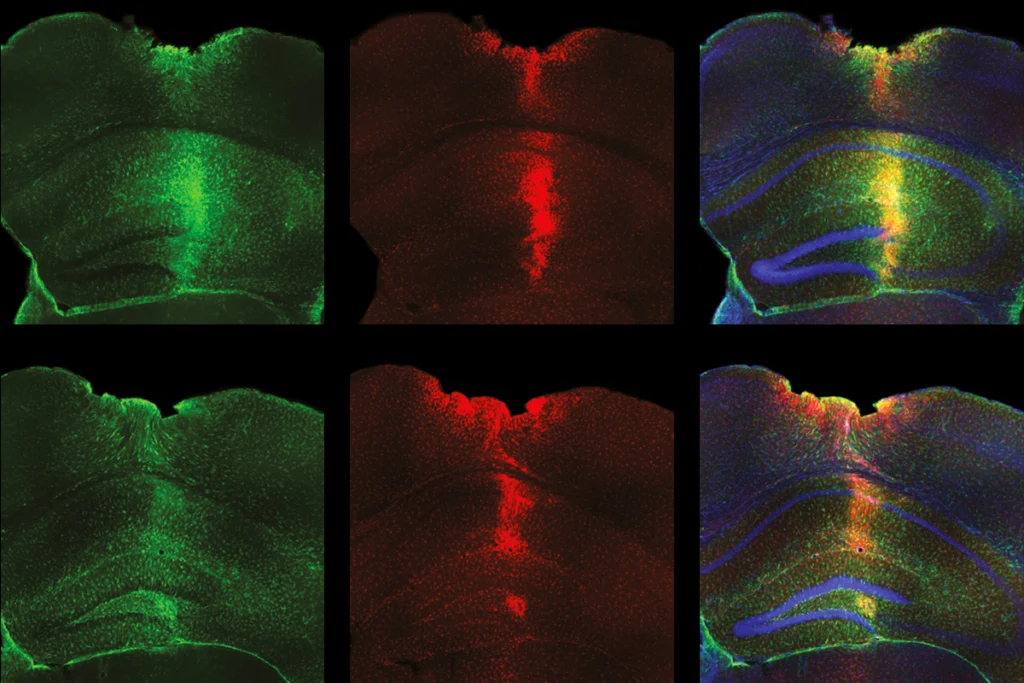
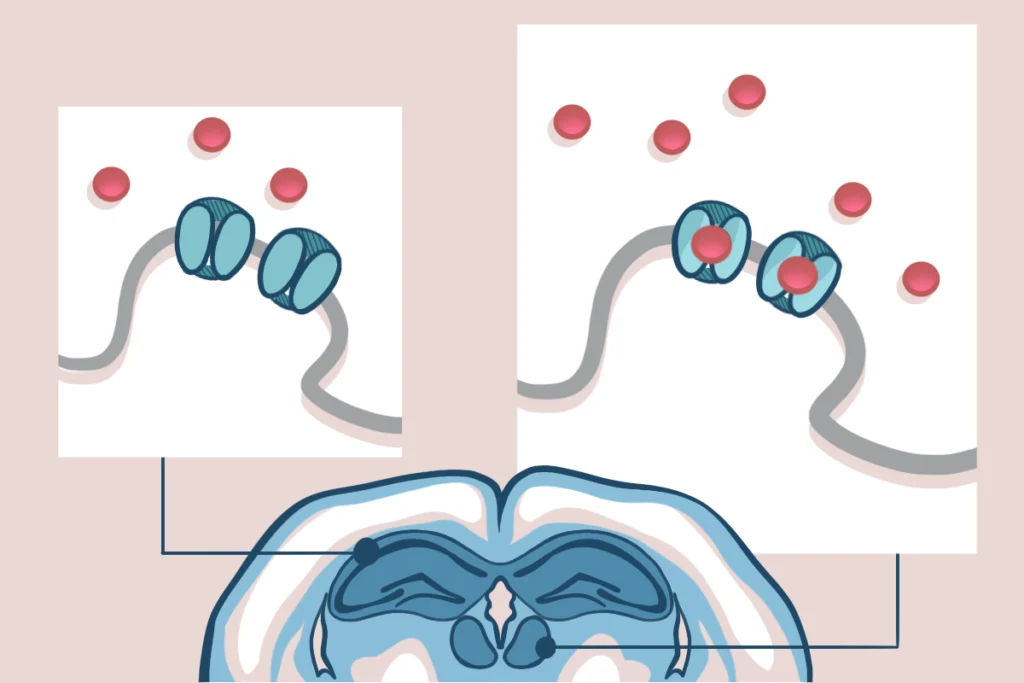
An alternative strategy is to focus on sexually dimorphic features in the brain, following the logic that these must interact with the neurobiology of autism. These interactions could occur at the level of the genome — if autism risk factors are expressed in a sexually dimorphic manner in the brain, for example. Alternatively, they could show up in downstream biological processes: For instance, components of the synapse that are involved in autism might be sexually dimorphic. The interaction may be transient, occurring at a specific stage of development, and specific to a single brain region, complicating its discovery.
At present, the FPE remains the most compelling explanation for the sex bias observed in autism, with strong supporting evidence from the observation of an excess of de novo mutations in girls with autism compared with boys. However, the absence of a clear difference in autism recurrence rate between the siblings of affected boys and affected girls poses an unsolved conundrum. As the genomic evidence for the FPE increases, the next challenge lies in attempting to leverage these findings to explore the nature of this protection.
Stephan Sanders is assistant professor of psychiatry at the University of California, San Francisco.