A novel gene therapy increases life expectancy and limits seizures in a mouse model of Dravet syndrome by selectively targeting inhibitory neurons, a new study shows.
The gene SCN1A codes for a type of sodium channel essential to the function of inhibitory interneurons. Mutations in SCN1A affect the electrical signaling of those cells and can result in Dravet syndrome, a severe form of epilepsy. About one-quarter of people with Dravet syndrome also have autism, and nearly half die prematurely from their seizures.
Because SCN1A is too large to fit into a typical adeno-associated virus (AAV), the standard delivery system for gene replacement therapies, many researchers have been working to instead ramp up the gene’s expression levels to compensate for the faulty copy. For instance, doing so with synthetic compounds called antisense oligonucleotides decreases the frequency of seizures and prolongs the lives of mice that model Dravet syndrome, according to a 2020 study.
In the new work, researchers outfitted an AAV with the gene for a transcription factor that boosts SCN1A expression, along with a regulatory element specific to inhibitory interneurons to restrict the therapy’s effects to those cells.
That selectivity potentially limits off-target effects, says lead investigator Stephanie Tagliatela, co-founder and chief scientific officer of Encoded Therapeutics, a gene therapy company in South San Francisco, California.
Unlike with excitatory neurons, specifically targeting inhibitory interneurons using AAVs “historically has been very difficult,” says Yang Yang, assistant professor of medicinal chemistry and molecular pharmacology at Purdue University in West Lafayette, Indiana, who was not involved in the work.
By combining gene therapy tools, the team was able to achieve exciting results, says Fyodor Urnov, professor of genetics, genomics and development at the University of California, Berkeley and director for technology and translation at the Innovative Genomics Institute. Urnov is also co-founder of Tune Therapeutics, another gene therapy company working on dialing levels of gene expression up or down.
“To get to the moon, you had to have woven together innovation from multiple, distinct, non-overlapping technologies,” Urnov says.
T
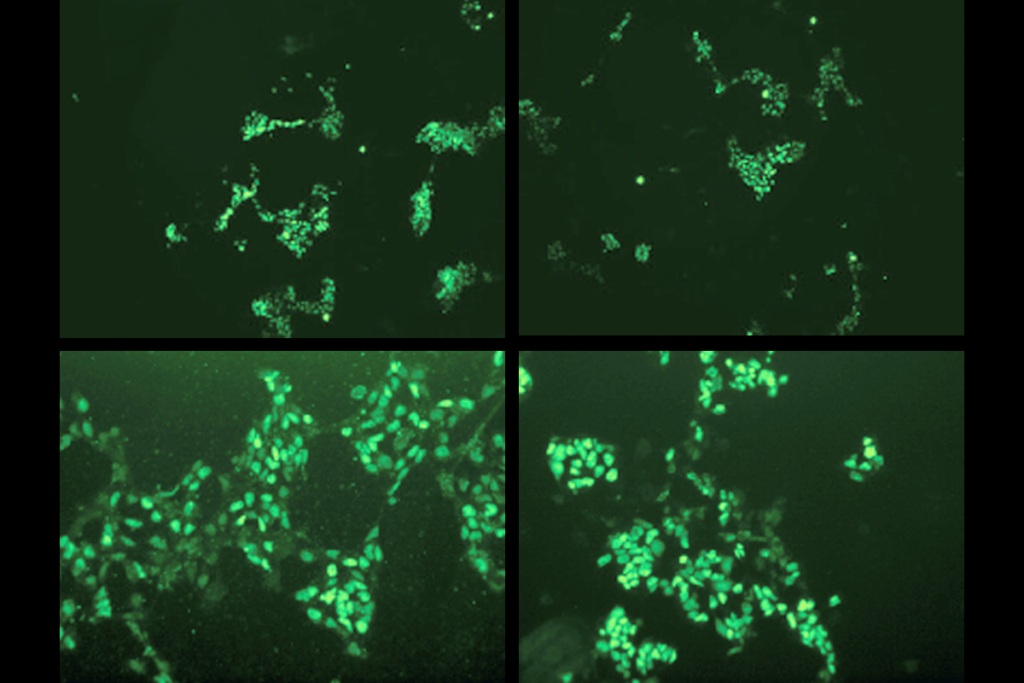
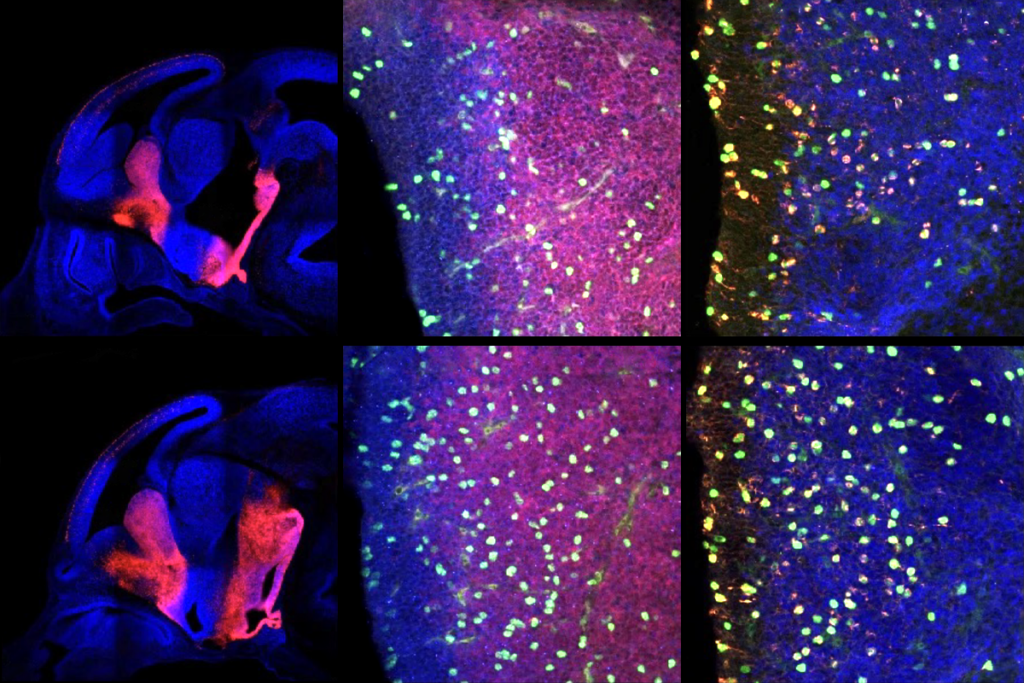
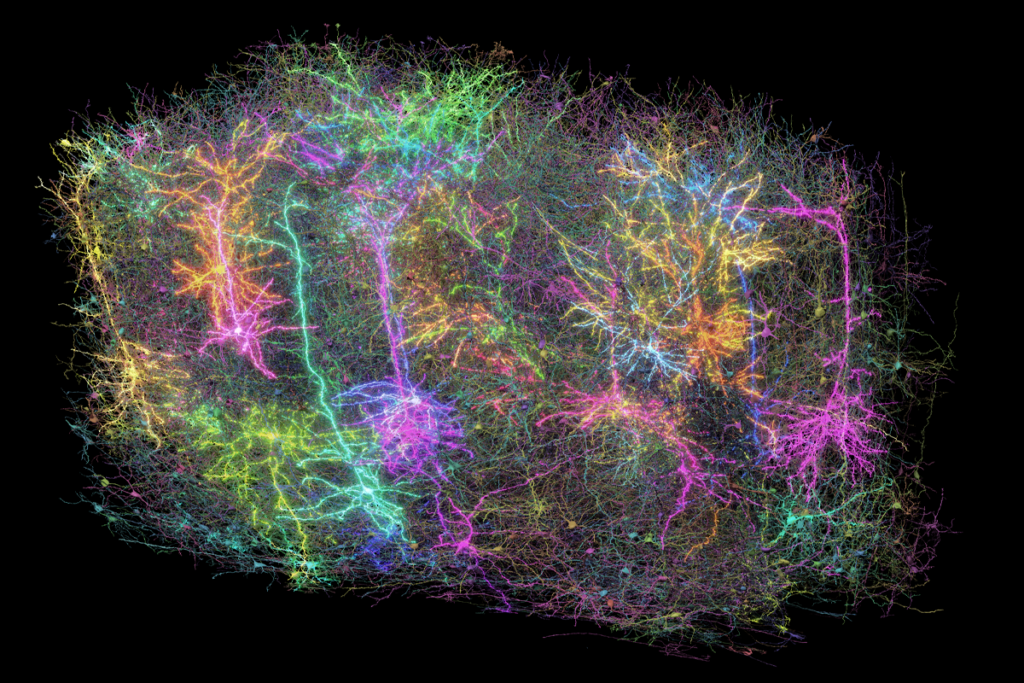
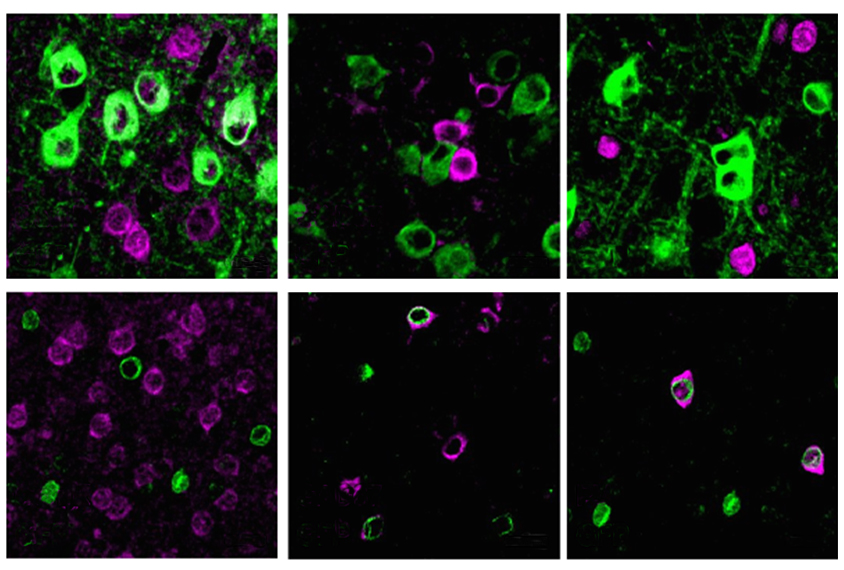
agliatela and her colleagues injected the modified virus, ETX101, into the right and left brain ventricles of 1-day-old Dravet syndrome model mice. The treated animals survived longer than those that received sham injections: 83.2 percent were alive more than one year later, compared with 31.4 percent of controls. The mice also had fewer and less severe seizures during a nine-day assessment period in the month after treatment.The virus preferentially targeted inhibitory neurons in the animals’ brain, the researchers discovered after staining postmortem tissue for cell-specific markers. And boosting SCN1A expression in those cells resulted in an increase in the appropriate sodium channels, the team found. The results were published in Human Gene Therapy in April.
Young cynomolgus macaque monkeys that received a single ventricular injection of ETX101 showed no negative side effects up to 28 days after treatment, the team found. The virus infected cells in multiple regions of animals’ brain, according to postmortem analysis of the tissue, suggesting that a single injection has widespread effects. And although the team detected the transcription factor in non-brain tissue, such as the animals’ liver and spinal cord, its levels were, respectively, 200 and 8 times lower than in the brain.
Tagliatela and her team at Encoded Therapeutics are working toward a clinical trial of ETX101 in people with Dravet syndrome. To that end, they plan to initiate a trial to study how the condition progresses during the first years of a child’s life — from increased seizures to effects on motor function — which will help them gauge the treatment’s safety and effectiveness, Tagliatela says.
“They’re going to have to show the FDA a much longer time point,” Urnov says. “But the fact that the monkey[s] did not keel over is tremendous news.”
B
ut even as the company eyes clinical trials, many questions remain. For one, it is unclear how treatment given later than the first few days of life would have affected the model mice, Yang says. If the therapy is given on day 10 or 20, he says, “can we still increase survivability?” Although it may be preferable to treat children with Dravet syndrome during their first day of life, most children are not diagnosed until they are at least a few months old, he says.Figuring out the window for intervention, the correct dose of treatment and the right way to treat children at different ages will be critical next steps, Yang adds.
“What they’re doing is very hard,” Urnov says. “The unmet medical need is enormous, and the disease is devastating. And every bit of data in this work bodes well for the fact that they’re going to be able to go to a first-in-human clinical trial,” in which children with Dravet syndrome could receive the treatment.
Still, he adds, “no amount of preclinical data can predict success. Things fail in human beings for all reasons — for reasons we don’t understand.”
Urnov likens the successful results in a mouse model to rowing across the Finger Lakes in New York. In that case, he says, “then doing a nonhuman primate experiment is sailing from Long Island to Cape Cod. And doing a human clinical trial is circumnavigating the globe.”