This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
A virus that ferries healthy copies of the Rett syndrome gene across the blood-brain barrier can reverse symptoms in female mice that model the disorder, according to a report published 21 August in the Journal of Neuroscience. The approach is the closest yet to simulating a workable treatment for the autism-related disorder.
A virus that ferries healthy copies of the Rett syndrome gene across the blood-brain barrier can reverse symptoms in female mice that model the disorder, according to a report published 21 August in the Journal of Neuroscience1. The approach is the closest yet to simulating a workable treatment for the autism-related disorder.
The first inkling of a way to treat Rett syndrome surfaced in 1999 when researchers traced its origin to mutations in MeCP22. Since then, studies have pieced together some of the gene’s many functions — including control of the expression of thousands of other genes.
For their study, the researchers began with female mice that have a single working copy of MeCP2. When placed on a new surface, these mice freeze or only hesitantly explore their environment, drag their hind legs when walking and tuck their legs when lifted instead of splaying them. They have tremors and irregular breathing, and generally look unkempt, with squinty eyes and ungroomed coats.
The researchers attached a copy of MeCP2 along with a piece of its promoter, or ‘on switch,’ to a viral Trojan horse. They then injected the virus into the blood of female mice aged 10 to 12 months. As they had hoped, the virus crossed into the brains of the mice and began expressing the MeCP2 protein in their brain cells.
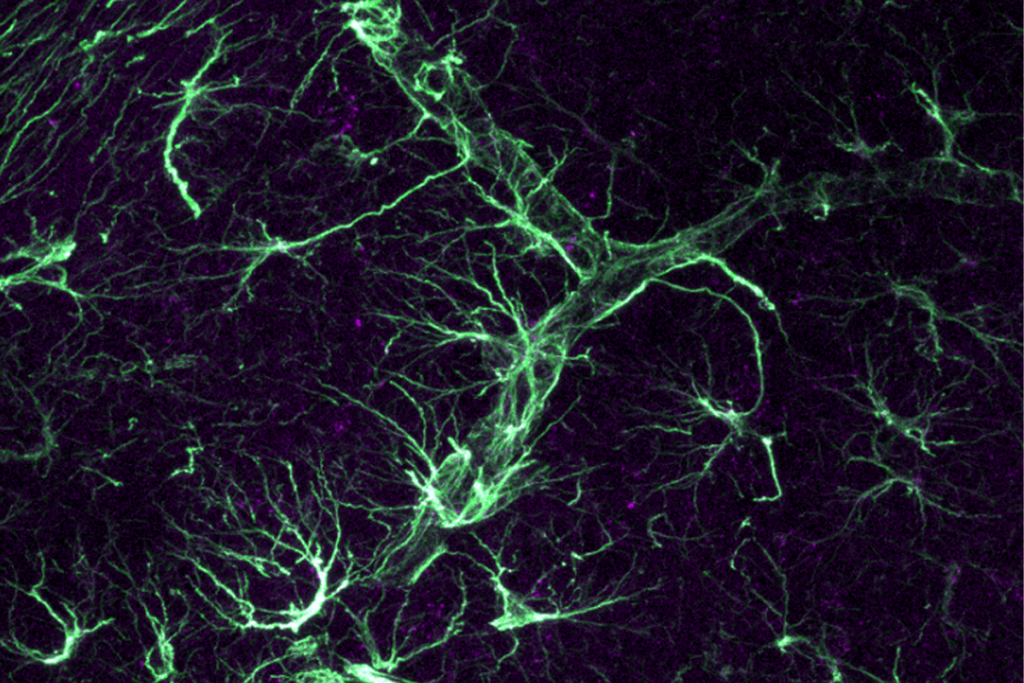
The researchers showed that the protein successfully bound to the DNA of 15 to 30 percent of the neurons and astrocytes — star-shaped cells that support neurons — in the brains of the mice. With an intact copy of the gene restored, within four to five weeks the mice began to lose their telltale symptoms and look more normal.
“You could actually look at the mice and see that they were healthier, more robust; they could make nests and so on,” says lead investigator Gail Mandel, professor of biochemistry and molecular biology at Oregon Health and Science University in Portland.
“What I was most surprised at was the amount of recovery we saw based on what I would call suboptimal expression in the brain,” Mandel says, “so it may be that in females that we don’t have to fix every cell.”
By 12 weeks after injection, the mice had regained most of their motor abilities, including how quickly and how far they moved around the cage, and how well they balanced on a beam.
These improvements held steady for at least six months after treatment, when the behavioral tests were complete. However, their respiratory problems did not show consistent improvement.
The rule of thumb for treating neurodevelopmental disorders has been the earlier the better, but Rett seems to be an exception. This therapy’s success in fully mature mice with pronounced symptoms has raised hopes of slowing or halting the progression of Rett syndrome even in older girls and women with the disorder.
Gene therapy has a troubled history, with reports of spectacular failures and, in rare cases, death. The approach first showed promise for treating neurological disorders in 2009, when researchers developed a viral vector called AAV9 that could cross the blood-brain barrier3.
A lab member of Mandel’s stumbled across this research at a meeting and instantly saw the potential for treating Rett syndrome.
Although she was hesitant at first to wade into unfamiliar work with a virus vector, Mandel says she realized Rett is a prime candidate for gene therapy for at least two reasons: Unlike autism, most cases of Rett are caused by mutations in a single gene located on the X chromosome. And because MeCP2 is a master regulator of other genes, treating any one aspect of the disorder with a drug could miss multiple other problems.
These reasons also help to explain why no single therapy has proven broadly effective for the disorder. Gene therapy, in contrast, zeroes in on the root of the problem, correcting the mutated gene.
What’s more, says Mandel, even in mature mice modeling Rett, neurons don’t show signs of irreversible damage.
“[This study] is quite promising on a number of levels,” says Stuart Cobb, senior lecturer at the University of Glasgow, who was not involved in the study. Last year, Cobb and his colleagues showed that AAV9 carrying MeCP2 can alleviate severe Rett-like symptoms and prolong survival in male mice aged 9 to 16 weeks when injected directly into the brain4.
“We were showing one could delay the onset of a phenotype and delay gross measures, whereas [Mandel’s] studies show that there’s potential for reversing established phenotypes,” says Cobb. “What’s required now are studies to optimize the vector and the route of delivery.”
Although Rett primarily affects girls, most research on the syndrome relies on male mice lacking MeCP2. That’s because male mice show symptoms earlier, develop more severe symptoms and die young, giving researchers a quick and easy read on the disorder.
For the same reasons, it’s also more challenging to treat males than it is females. Developing a gene therapy for females has its own pitfalls, however. Girls have two X chromosomes — one that carries the damaged gene and the other intact. In any given cell, only one of the chromosomes is turned on, meaning that only about half the cells in girls with Rett syndrome have a mutant copy of the gene.
“It is a tricky disorder in that not all the cells are sick; half of them are healthy,” says Lucas Pozzo-Miller, professor of neurobiology at the University of Alabama, Birmingham, who was not involved in the study.
“The concern was that in replacing the MeCP2 for the cells that need it, one could also disrupt the MeCP2 in the cells that were functioning normally,” causing it to be doubly expressed, he says. This could cause problems like those seen in MeCP2 duplication syndrome, a rare condition with Rett-like symptoms.
Still, the new study suggests that gene therapy is an approach worth pursuing for Rett syndrome, Pozzo-Miller says. “Ideas are very cheap in biology. The success comes from sitting down and actually doing it,” he says. “In this case, it’s raising the hope that gene therapy is actually viable.”