Mice missing an autism-linked chunk of chromosome 16 have impaired neuronal movement during embryonic development and reduced cortical volume, a new study shows. But boosting expression of just one of the missing genes, TAOK2, can restore typical brain-cell migration in the animals.
TAOK2 is one of 29 genes on a segment of chromosome 16 called 16p11.2. People missing this region often have an enlarged head and developmental delay, and about 30 percent also have autism. People with mutations in one copy of TAOK2 may also have autism traits.
“In the field, a big question has been whether we can narrow all these genes [on 16p11.2] to a few that might be contributing to [autism],” says Smita Yadav, assistant professor of pharmacology at the University of Washington in Seattle, who was not involved in the work. It’s exciting to connect TAOK2 to a particular phenotype, she adds.
TAOK2 encodes two protein variants, or isoforms: alpha and beta. Beta helps with the formation of a neuron’s dendritic spines, according to a previous study by the same group.
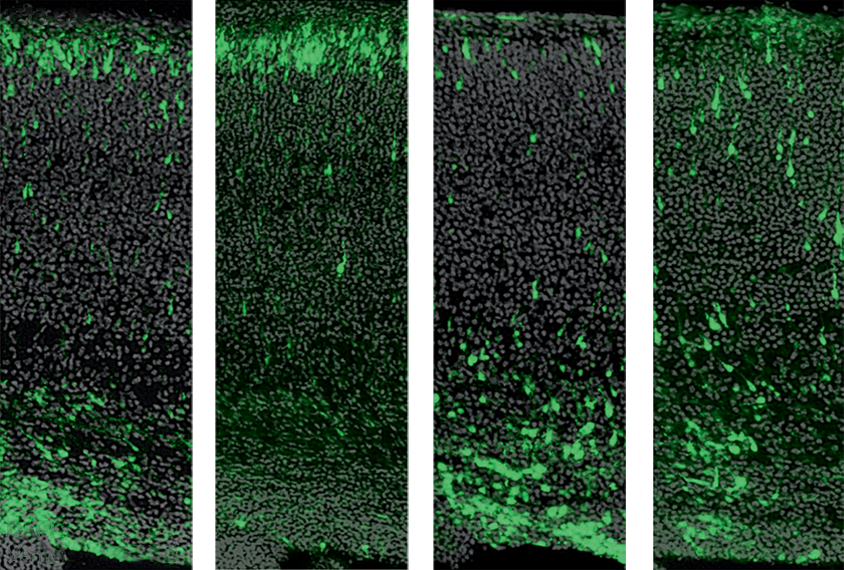
The alpha version of TAOK2 controls neuronal migration, according to the new work. The researchers introduced autism-linked mutations to impair either the alpha or beta isoform in prenatal mice. Four days later, an analysis of brain tissue samples revealed altered cell positioning in the cortices of alpha-mutated animals only.
“TAOK2 is implicated in different aspects of neural development,” says lead researcher Froylan Calderón de Anda, head of the neuronal development research group at the University Medical Centre Hamburg-Eppendorf in Germany. “Depending on the isoform, you’ll get different effects.”
The two isoforms add a plot twist to the story, says Santhosh Girirajan, assistant professor of biochemistry and molecular biology at Pennsylvania State University in University Park, Pennsylvania, who was not involved in the study. “It’s like watching a movie where you think there’s one person, but it’s actually twins.”
E
nhanced expression of both isoforms revealed that only alpha binds to microtubules, components of a cell’s internal scaffolding that are essential for its movement. JNK1, an enzyme that stabilizes microtubules, was less active in TAOK2-deficient mice, whereas neuronal migration resumed after the introduction of an activated version of JNK1.Defective neuronal migration may be one reason that people with 16p11.2 deletion syndrome have a thinner cerebral cortex, says study investigator Melanie Richter, a postdoctoral researcher in de Anda’s lab. “The cells end up in the wrong layer and do not connect with the correct targets.” Those cells are not active enough, Richter says, and end up dying as a result.
The team compared the TAOK2-deficient mice with animals missing one copy of 16p11.2 and found similar migration defects, which were normalized by turning up expression of TAOK2 alpha.
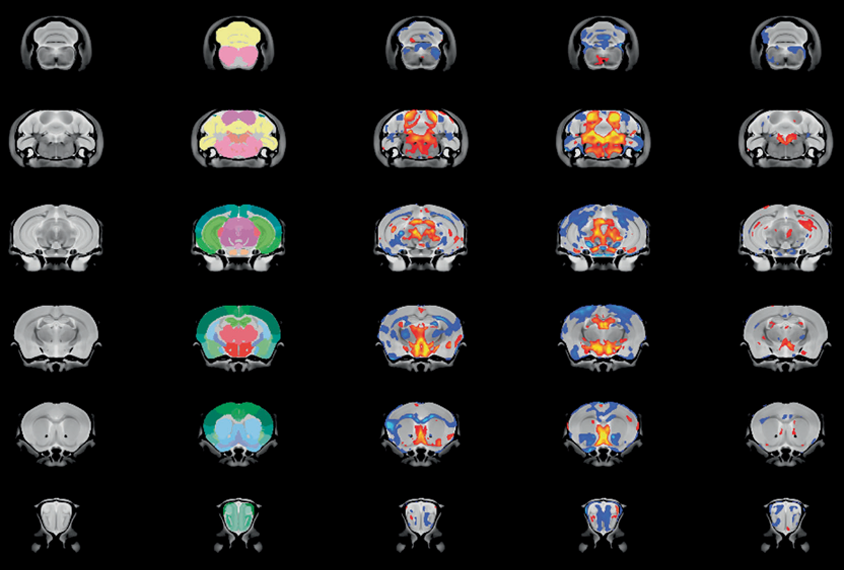
MRI also revealed similarities in brain anatomy, including cortex volume, between the two models. The 16p11.2 deletion mice shared less resemblance with mice lacking KCTD13, another autism-linked gene in the same 16p chromosomal region.
The findings were published on 19 September in Molecular Psychiatry.
The results point to TAOK2 as a main player in 16p11.2-linked autism and an important therapeutic target, Richter says. “It is really the only known contributor to the 16p-associated phenotype,” she adds.
Neuronal migration and cortex size are only two aspects of the 16p phenotype, however, Girirajan says, noting that other genes in the 16p region likely contribute to other aspects of the phenotype.
The team continues to dissect the roles of the TAOK2 isoforms. Unpublished data from the group suggest that TAOK2 beta regulates protein production in neurons.
“Now we are trying to do a more general approach using a protein synthesis inhibitor in prenatal animals to see if we can change their behavioral issues,” says de Anda.