Mice with a mutation that boosts the activity of the autism-linked protein UBE3A show an array of behaviors reminiscent of the condition, a new study finds. The behaviors differ depending on whether the animals inherit the mutation from their mother or their father, the work also reveals.
The results add to mounting evidence that hyperactive UBE3A leads to autism. Duplications of the chromosomal region that includes UBE3A have been associated with autism, whereas deletions and mutations that destroy the gene’s function are known to cause Angelman syndrome, which is characterized by developmental delay, seizures, lack of speech, a cheerful demeanor and, often, autism.
“UBE3A is on a lot of clinicians’ radar because it is well known to be causative for Angelman syndrome when mutated or deleted,” says lead investigator Mark Zylka, professor of cell biology and physiology at the University of North Carolina at Chapel Hill. “What our study shows is that just because you have a mutation in UBE3A, it doesn’t mean that it’s going to be Angelman syndrome.”
In the cell, UBE3A is involved in the degradation of proteins, and “gain-of-function” mutations — which send the UBE3A protein into overdrive — result in enhanced degradation of its targets, including UBE3A itself. Studying the effects of these mutations could provide insight into how they affect brain development and suggest targets for therapies, says study investigator Jason Yi, assistant professor of neuroscience at Washington University in St. Louis, Missouri.
G
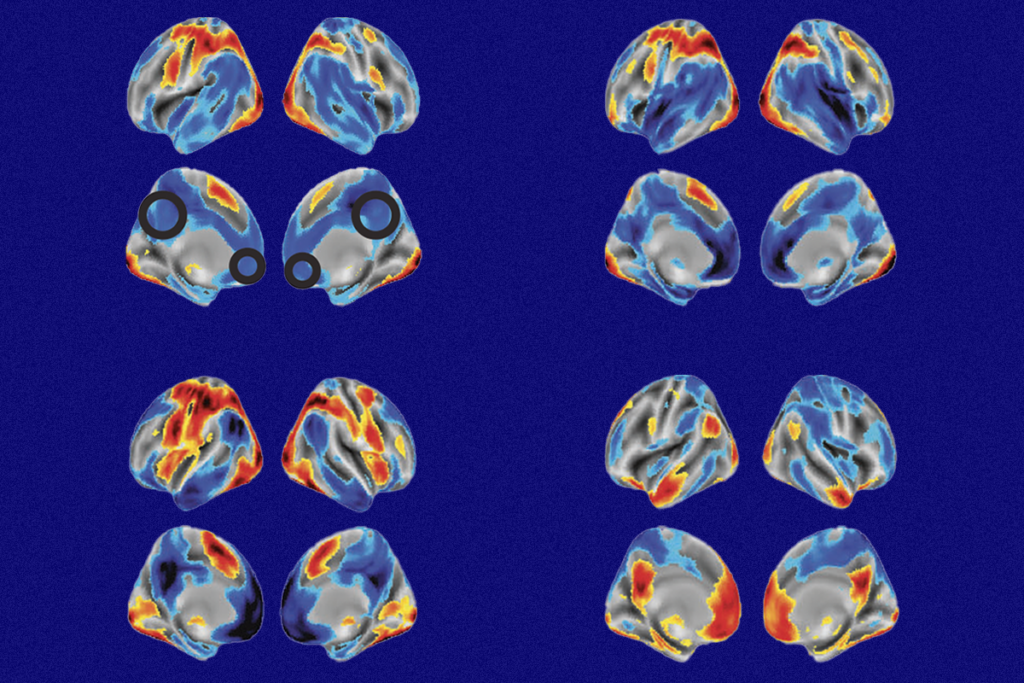
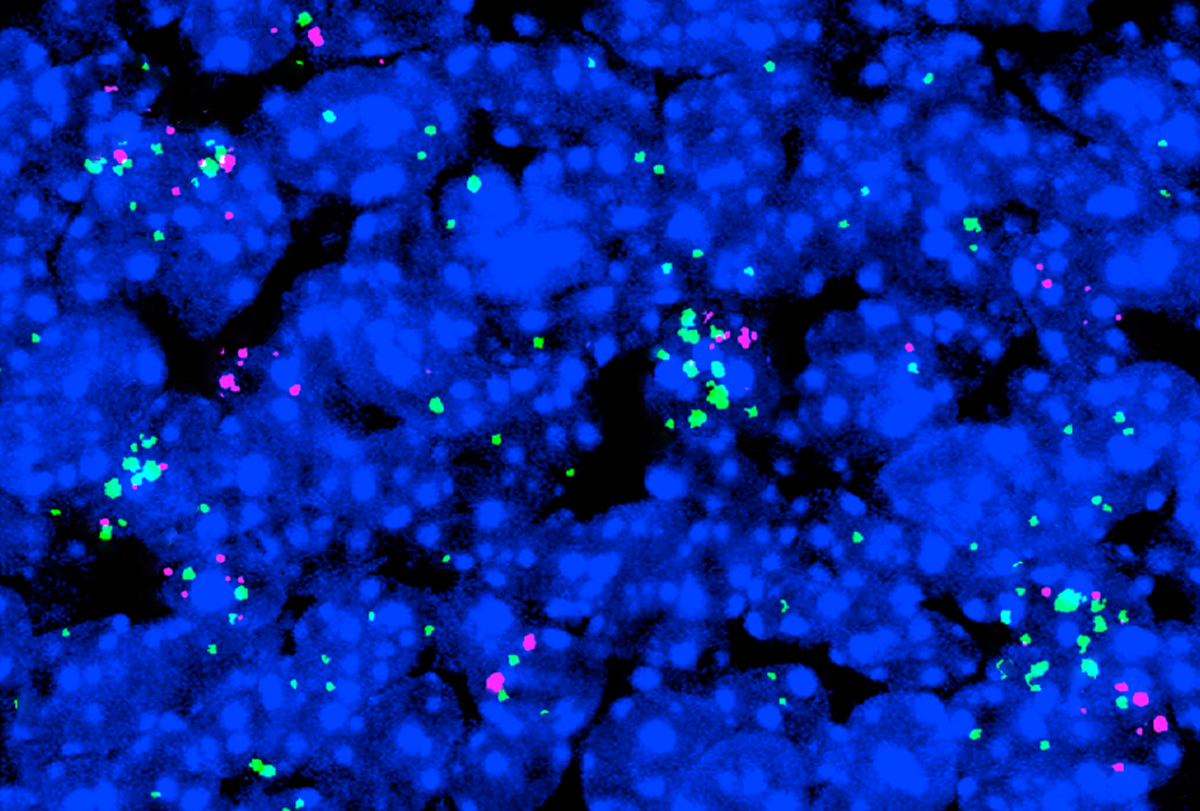
ain-of-function mutations in UBE3A can disrupt early brain development and may contribute to neurodevelopmental conditions that are distinct from Angelman syndrome, Yi and Zylka have shown in previous studies. One of the mutations they analyzed had been found in an autistic child, so the team used CRISPR to create mice with this mutation.During embryonic development, the mice show increased numbers of a specific subset of inhibitory interneurons in the brain’s outer layer compared with controls. Problems with interneurons can lead to the imbalance between neuronal excitation and inhibition seen in some forms of autism, previous studies suggest.
The differences in the number of interneurons mostly disappeared after birth, Zylka’s team found. The researchers now plan to investigate whether the alterations seen during embryonic development in mutant mice have a lasting effect on the imbalance of excitatory and inhibitory brain signals.
T
he parent of origin of UBE3A mutations can influence the severity of behavioral traits. This is because although people inherit two copies of the gene, only the one inherited from the mother is active in neurons — the paternal copy is silent. So Zylka’s team set out to study three groups of mutant mice: one that inherited the mutated copy of UBE3A from their mother, one that inherited it from their father, and one that inherited two mutated copies of UBE3A.Mice that inherited the mutated UBE3A from their mother and those that inherited two mutated copies, but not those that inherited the mutated UBE3A from their father, were hyperactive compared with controls. In a test that assesses sociability in mice, only animals that inherited the mutated UBE3A from their father failed to show a preference for an unfamiliar mouse versus an empty cage — a behavior indicative of social deficits.
In a test of motor coordination, in which mice are placed on a rod that spins at increasing speed, mutant mice performed similarly to controls. Mice that inherited the mutated UBE3A from their mother stayed on the rod even longer than control animals, the researchers found. This is in contrast with what has been observed in mouse models of Angelman syndrome, which fall off the rotating rod sooner than controls, Zylka says.
The researchers reported their results in Cell Reports last month.
T
he findings suggest that gain-of-function mutations in UBE3A result in an autism-like condition that is different from Angelman syndrome, Zylka says. “All the clinical data and this preclinical data point to this being a new syndromic disorder, with phenotypes that are going to vary by parent of origin and by the severity of the gain-of-function mutation.”The results may also have implications for clinical studies. Several companies are pursuing gene therapies for Angelman syndrome that restore typical UBE3A levels by unsilencing the paternal gene copy. But, Zylka notes, this approach may not work — and may even be counterproductive — in people who have hyperactive UBE3A.
The study indicates that overactive UBE3A in non-neuronal cell types, such as neuron-supporting glial cells, may be causing the autism traits found in some people with a gain-of-function mutation in this gene, says Lawrence Reiter, professor of neurology at the University of Tennessee Health Science Center in Memphis, who did not take part in this research. Unlike in neurons, which express only one copy of UBE3A, both copies of the gene are always active in non-neuronal cells, and Reiter’s research in fruit flies suggests that an excess of UBE3A expression in glia causes seizures. In people, a duplication of the chromosomal region that includes UBE3A results in a condition characterized by autism, seizures and cognitive problems.
“Glial cells have an influence on neuronal activity, and if you alter UBE3A levels or activity in glia, you get a phenotype,” Reiter says. “We should stop focusing so much on neurons for the [autism] phenotypes.”