This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
Problems with the minute ‘factories’ that yield energy for cells — the mitochondria — may contribute to learning problems and other traits of fragile X syndrome.
Problems with the minute ‘factories’ that yield energy for cells — the mitochondria — contribute to learning problems and other traits of fragile X syndrome, a new study suggests1. The findings also unexpectedly implicate huntingtin, the protein mutated in Huntington’s disease, in fragile X.
Fragile X syndrome is the most common inherited form of intellectual disability and single-gene cause of autism. It stems from mutations that silence the gene for a protein called FMRP. The absence of this protein alters the production of many others, with consequences trickling down to affect the form and function of neurons.
One such consequence, the new study suggests, is faulty, fragmented mitochondria.
Mitochondria fuse to create larger units during times of high energy demand, such as cell growth. This mitochondrial fusion is decreased in neurons deprived of FMRP, the researchers found. Enhancing the fusion in a mouse model of fragile X normalizes neurons’ form and function, and improves the mice’s learning and behavior.
“This is a new mechanism, and I think it’s only a part of the bigger picture of what happens in fragile X,” says lead investigator Xinyu Zhao, professor of neuroscience at the University of Wisconsin-Madison. But it gives scientists a clear therapeutic target, she says. “It’s very exciting because it suggests we don’t have to touch every single knob in order to treat the symptoms.”
Researchers have for years attempted to find treatments for fragile X. But many candidates that showed promise in animals failed in people. As a result, researchers are seeking new targets to explore.
“This is an exciting new avenue to be pursued for development of therapeutics,” says Mark Bear, professor of neuroscience at the Massachusetts Institute of Technology, who was not involved in the study.
In a 2015 study, Zhao and her colleagues found that FMRP is important for newborn neurons in the mouse hippocampus, a hub for learning and memory; mice missing FMRP have problems with both2.
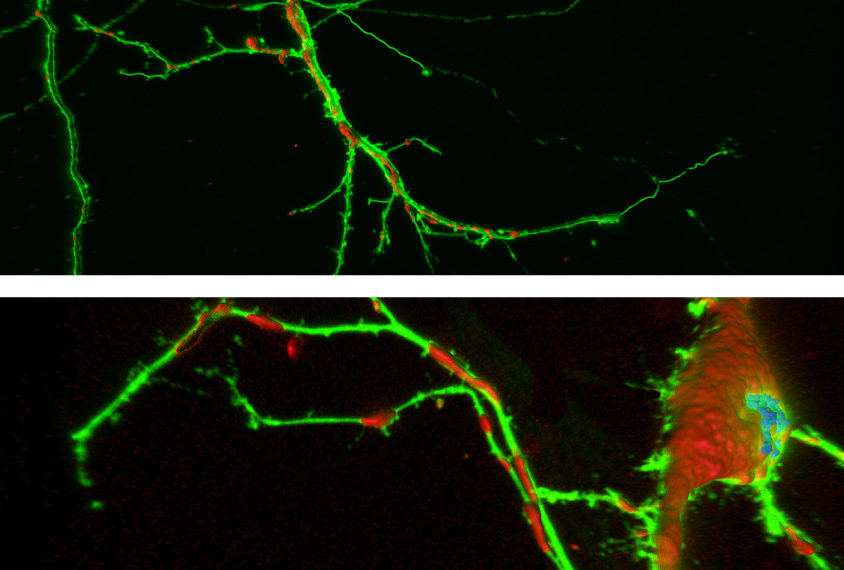
Mouse neurons lacking FMRP don’t mature properly. They have shorter and fewer dendrites — the branch-like protrusions that receive signals from other cells — than usual, the team found in the new study.
The researchers saw the same stunted dendrites in neurons from people with fragile X after transplanting immature versions of the cells into the brains of live mice. They also saw another telling defect: The transplanted cells show high levels of a marker for oxidative stress, a sign of metabolic problems.
To find the source of the problems, the researchers analyzed gene expression in the neurons. They found unusually low expression of two genes that induce mitochondria to fuse.
Treating the neurons with a compound called M1, which promotes mitochondrial fusion, normalizes the dendrites. (The drug has no effect on typical neurons.) Given to mice lacking FMRP, M1 improves the animals’ learning and memory and social interaction, and it eases their hyperactivity.
The merged energy factories may be needed for the maturation of dendrites, says Elizabeth Jonas, professor of internal medicine and neuroscience at Yale University, who wasn’t involved in the study.
FMRP does not seem to directly regulate mitochondrial fusion, Zhao says; none of the 40 common targets of FMRP are among the 519 genes with altered expression in fragile X mouse neurons.
But a database of protein interactions revealed that 18 of the 40 target genes interact with some of the 519 genes. One interacts with mitochondria-related genes exclusively: huntingtin, which underlies the degenerative condition Huntington’s disease.
“We just stumbled on huntingtin, which was a big surprise,” Zhao says.
People with Huntington’s disease produce an abnormally long version of huntingtin. The protein is of normal length in the fragile X mice, but there is too little of it. Boosting the protein’s levels restores mitochondrial function in fragile X neurons from mice; it also normalizes the dendrites.
“This provides support that huntingtin is actually a mediator of FMRP’s regulation of mitochondria,” Zhao says. The work appeared 11 February in Nature Neuroscience.
The role of mitochondrial functioning in brain development is poorly understood but gaining attention. Several conditions, such as Rett and Down syndromes, are linked to problems in mitochondria, and scientists have identified genetic changes that affect mitochondria in people with autism.
In fragile X research, too, mitochondrial function is an emerging new direction, Jonas says. In her unpublished work, she has found another problem with mitochondria in fragile X that leads to inefficient energy production, protein overproduction at synapses — the junctions between neurons — and poor maturation of neurons.
Zhao’s team, meanwhile, is exploring dietary supplements and medications that enhance mitochondrial function as possible treatments for fragile X syndrome.