This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
A new study may help explain why the symptoms of Rett syndrome appear 6 to 18 months after birth. It suggests that the role of MeCP2, the protein missing in the syndrome, becomes more important as the brain matures.
The signs of Rett syndrome typically emerge between 6 and 18 months of age, when girls who appear to be developing normally start losing their language and motor skills.A new study, published 28 April in the Proceedings of the National Academy of Sciences, may help explain this sudden turn. It suggests that the role of MeCP2, the protein missing in Rett syndrome, becomes more important as the brain matures1.
MeCP2 is known to regulate the expression of many other genes, dialing their expression up or down. Previous studies have shown that MeCP2 binds to DNA marked by a chemical tag called methyl CG (mCG). The new study shows that MeCP2 binds to three additional types of methylation sites, all of which accumulate after birth.
“If the docking sites that are really important for the gene expression changes are not even there early on, you can see why you don’t see Rett much earlier on,” says lead researcher Huda Zoghbi, professor of genetics, pediatrics and neuroscience at Baylor College of Medicine in Houston, Texas.
As these methylation sites build up, the effects of MeCP2 deficiency may become more pronounced, spurring symptoms of Rett syndrome.
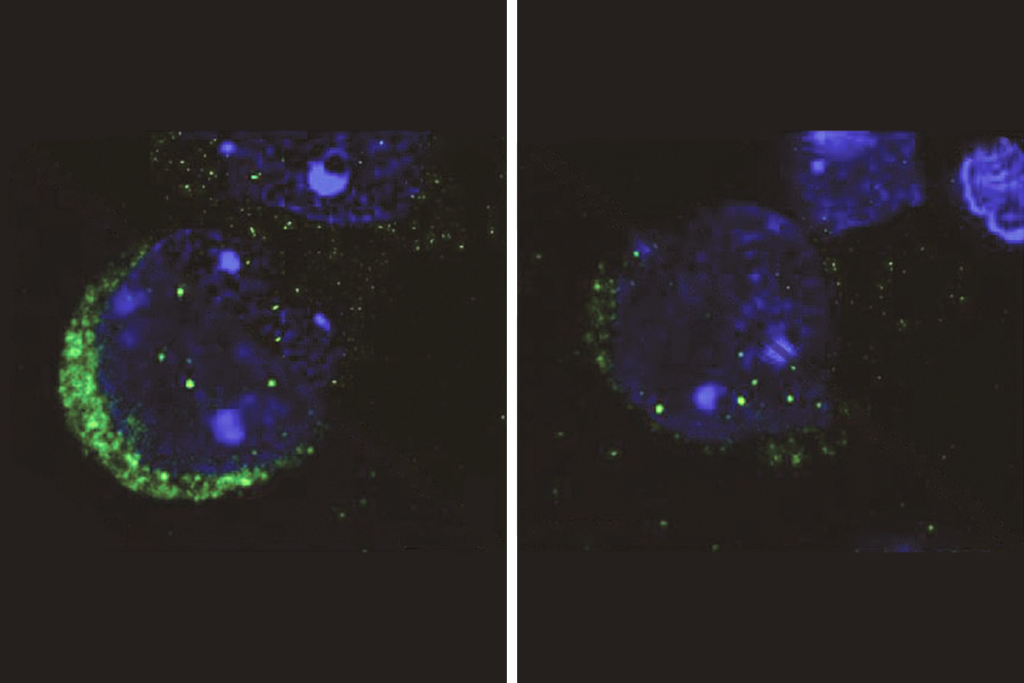
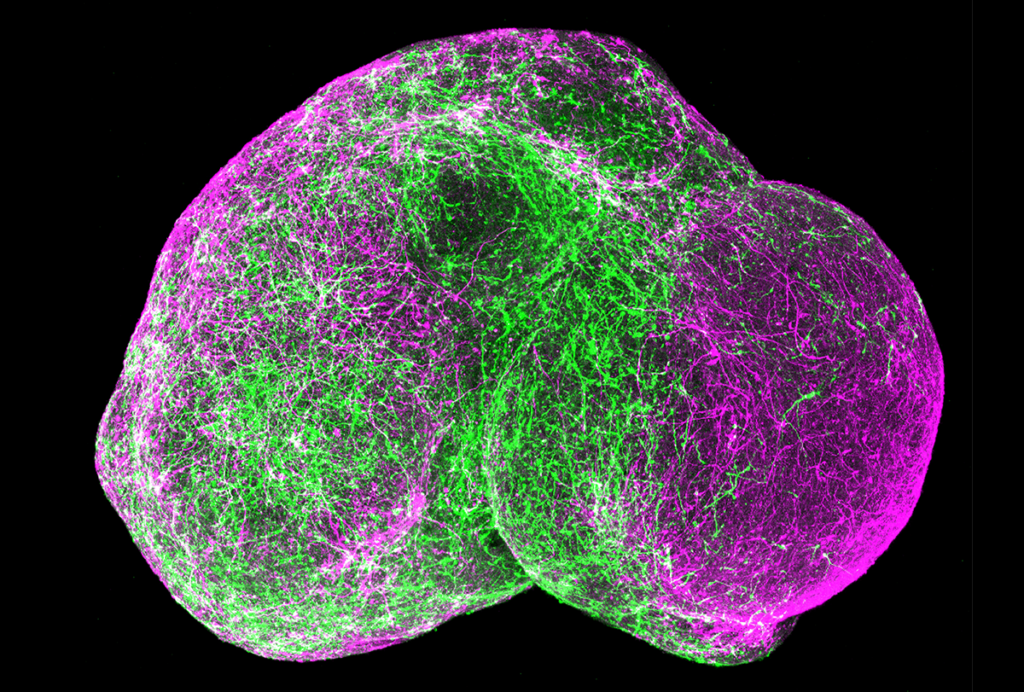
Zoghbi and her colleagues used brain tissue from mice to study how MeCP2 interacts with different sequences of DNA. As expected, they found that the protein binds to mCG sites. But it also binds to three other types of tags, dubbed mCA, mCT and mCC. These three sites are commonly known, collectively, as mCH.
The team then looked at mice missing MeCP2 — a model for Rett syndrome — and found that the genes that are disrupted are dense with mCH sites. By contrast, there is no association between the disruption of these genes and the density of mCG sites. This suggests that MeCP2 alters gene expression through interactions with mCH, not mCG.
Intriguingly, mCH sites are known to proliferate as the brain matures. The number of mCG sites, on the other hand, remains relatively constant. This accumulation of mCH sites over time could explain why the symptoms of Rett take months to appear, Zoghbi says.
The findings add to mounting evidence that methylation sites other than mCG play an important role in Rett syndrome, says Gail Mandel, senior scientist at the Vollum Institute at Oregon Health & Science University. A study published earlier this year found that MeCP2 regulates the expression of unusually long genes that are critical for brain function by binding to mCA sites.
“It is reassuring that several labs now, using different approaches, have arrived at similar conclusions,” says Mandel, who was not involved with either study.
1: Chen L. et al. Proc. Natl. Acad. Sci. USA 112, 5509-5514 (2015) PubMed