Neurons derived from people with 22q11.2 deletion syndrome, a genetic condition linked to autism, show deficits in electrical activity and calcium signaling, according to a new study1. A single gene appears to be largely responsible for these defects, the study shows.
Up to 20 percent of people who lack part or all of the chromosomal region 22q11.2 have autism. Individuals with the deletion may also have schizophrenia, seizures, heart defects, immune dysfunction or learning problems.
The new findings uncover factors that may contribute to the development of psychiatric conditions associated with 22q11.2 deletion syndrome. They could also help researchers identify new therapeutic targets, says lead author Sergiu Pasca, associate professor of psychiatry and behavioral sciences at Stanford University in California.
The syndrome is relatively common, occurring in up to 1 in 4,000 newborns, Pasca says. But researchers do not fully understand how genes in the 22q11.2 region contribute to autism or other conditions, he adds.
Brain signature:
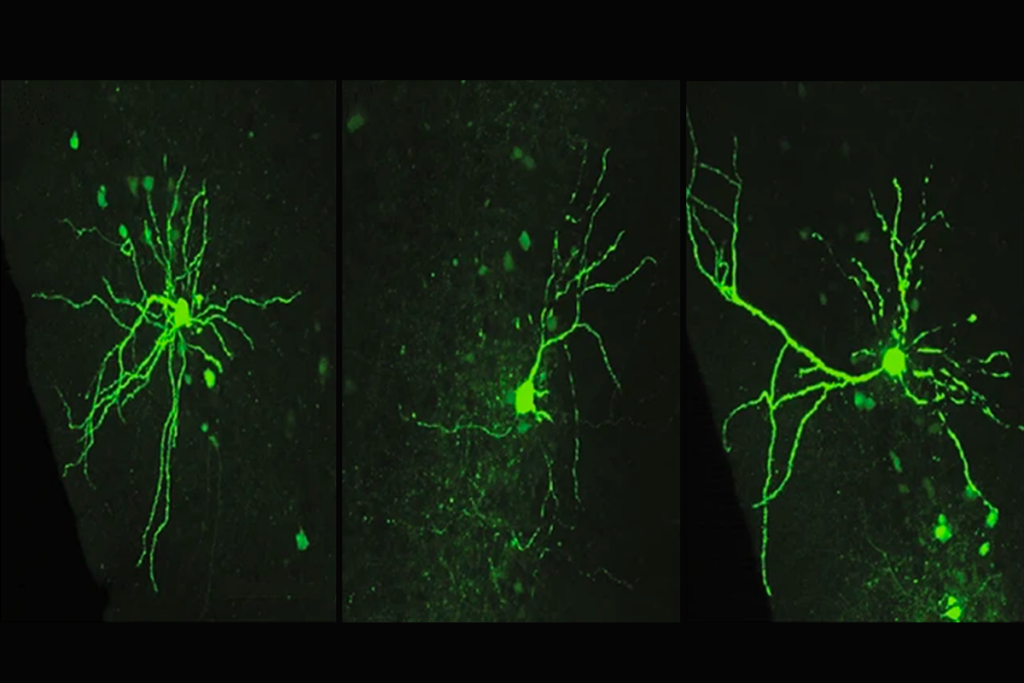
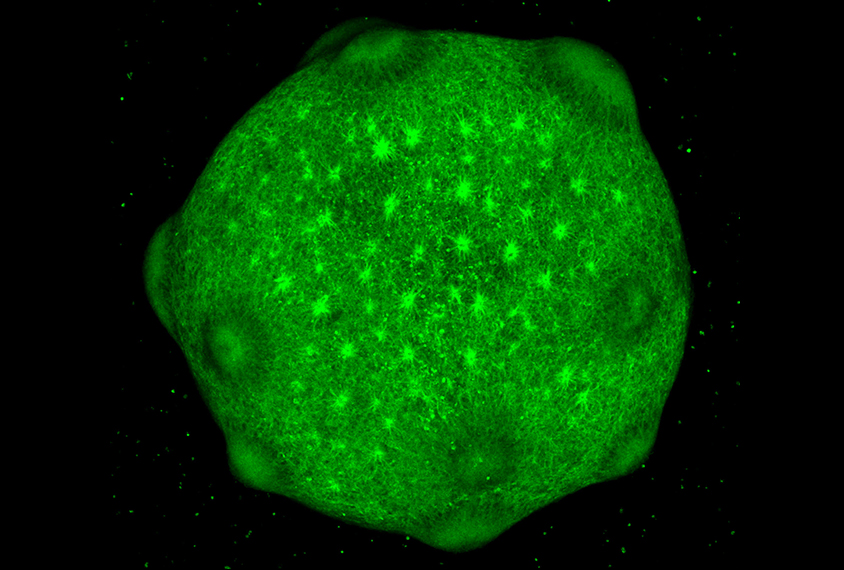
To solve this molecular puzzle, Pasca and his team reprogrammed skin cells from 15 people with the deletion and 15 controls into induced pluripotent stem cells. Using a technique they developed in 2015, they coaxed these cells to turn into neurons, which self-organize in a dish into spherical clusters called organoids. The organoids show some key features of the developing cerebral cortex, a brain region implicated in autism.
The neurons derived from people with 22q11.2 syndrome spontaneously fire four times as frequently as neurons derived from controls, the researchers found. And the electrical activity of the 22q cells does not set off the usual spike in calcium levels, which is crucial for neurons to exchange messages.
In some other syndromes tied to schizophrenia and autism, calcium-channel genes are mutated. But the number of channels and the speed at which they work in 22q neurons is the same as in control neurons. Instead, the 22q cells show an unusually low voltage difference across the cell membrane when they aren’t firing, causing the signaling defects and hyperexcitability, the researchers found.
The researchers suspected that a gene called DGCR8 might be responsible for the neuronal deficits in the organoids because it lies within 22q11.2 and is linked to abnormal electrical activity in the neurons of mice2. DGCR8 is essential for the synthesis of short RNA fragments, called microRNAs, that regulate gene expression.
Lowering DGCR8’s expression levels in control neurons reproduced the abnormalities seen in 22q neurons. In contrast, boosting the gene’s activity in 22q neurons or treating them with antipsychotic drugs prevented them from being overly excitable and reversed their calcium-signaling defects. The study was published 28 September in Nature Medicine.
Treatment target:
Previous studies have analyzed lab-grown neurons derived from people with schizophrenia or autism-related disorders such as Rett and fragile X syndromes. But most used only a few human-derived cell lines, says Guo-li Ming, professor of neuroscience at the University of Pennsylvania in Philadelphia. The new study, Ming says, “has a total of 30 human lines — that’s a huge effort.”
By studying brain organoids derived from so many people, the researchers were able to identify the gene that might be involved in the psychiatric conditions associated with 22q11.2 syndrome, says Sally Temple, scientific director of the Neural Stem Cell Institute in Rensselaer, New York. “Whenever we have a light shining ahead, saying, ‘This is what you should really be looking at,’ it means that we’re making progress,” she says.
The study participants with 22q11.2 syndrome vary in their psychiatric diagnoses, and yet all the brain organoids derived from their cells show the same neuronal abnormalities. “That’s somewhat surprising, because we know there are a lot of differences in the genetic background of different people,” Ming says.
The deletion might conspire with other factors to ultimately determine which psychiatric conditions a person has, Pasca says. “It could be that the deletion causes cellular defects, and once there is a stressor such as social stress, disease develops.” It’s also unclear whether these cellular defects are related to the high prevalence of seizures in people with 22q11.2 syndrome, he says.
The hallmarks of most neuropsychiatric conditions can change over time, says Giuseppe Testa, director of the stem cell epigenetics unit at the European Institute of Oncology in Milan, Italy. Studies that look at a larger number of people with 22q11.2 deletion syndrome or other neurodevelopmental conditions could help to elucidate the relationship between the neuronal defects observed in the lab and the psychiatric manifestations of the conditions, Testa says. The new study, however, is a “great resource” for understanding how the 22q11.2 deletion contributes to schizophrenia and autism, he says.
Pasca’s team is trying to pinpoint molecules that could open new therapeutic avenues for 22q11.2 deletion syndrome. The antipsychotics they tested restore the unusual voltage differences in the 22q neurons, but they don’t address the core mechanisms responsible for psychiatric conditions linked to the syndrome, Pasca says.
What’s more, antipsychotics have many side effects, and not all individuals respond to them, he says. “We need better therapies — we need to identify what the key molecular players are and target those.”