This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
Mutations in a gene linked to intellectual disability and sometimes autism may lead to a permanent boost in brain activity, according to a study published 18 June in Neuron.
Mutations in a gene linked to intellectual disability and sometimes autism may lead to a permanent boost in brain activity beginning early in development, according to a study published 18 June in Neuron1.
The gene, called SYNGAP1, prevents connections that send activating signals in the brain from maturing too early during the first few weeks of fetal development. Mutations that inactivate one of two SYNGAP1 gene copies are a relatively common cause of intellectual disability but also frequently lead to epilepsy and, in about 30 percent of cases, autism.
In 2012, the same researchers showed that loss of one copy of this gene results in abnormally strong signals and overexcitable brains in mice2. The boost in activating signals may lead to an imbalance between inhibitory and excitatory activity, which some researchers have suggested as a cause of autism and epilepsy.
The new study suggests that mutations in SYNGAP1 cause permanent damage to the brain in utero. Restoring SYNGAP1 in adulthood does not improve symptoms in mice missing a copy of the gene. Conversely, removing it in adulthood does little damage to their brains and behavior.
“You can lose one copy of SYNGAP1 in adulthood, that’s fine,” says lead researcher Gavin Rumbaugh, associate professor of neuroscience at the Scripps Research Institute in Jupiter, Florida. “But you need full expression of the protein in the developmental critical period in order to have cognition develop and emerge properly.”
The results may seem discouraging for developing treatments. But they suggest that repairing the mutation in utero may completely mitigate its effects. “If you had a compound that enhanced SYNGAP1 back up to normal in the critical period, you would essentially cure the disorder,” says Rumbaugh — although he notes that putting this strategy into practice is decades away.
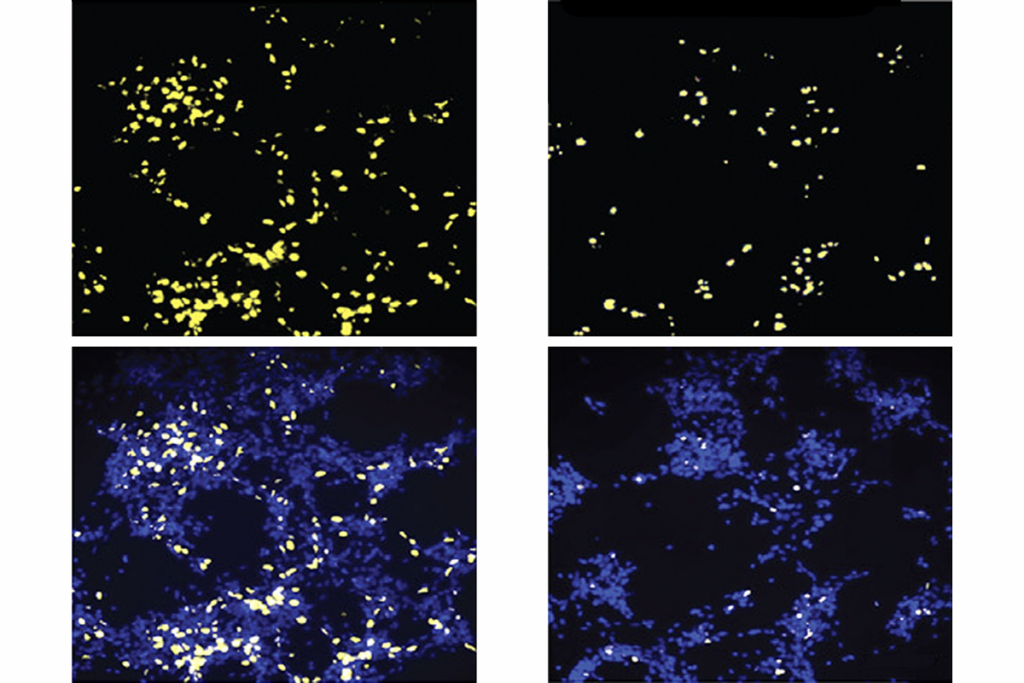
The study also looked at where in the brain a SYNGAP1 mutation might cause the most damage. Pyramidal neurons, which send excitatory signals in the forebrain — a part of the brain responsible for most cognitive function — are most affected.
“Understanding which neuron types are important will help us to understand mechanism and how these disease genes are working in specific neurons,” says Kimberly Huber, professor of neuroscience at the University of Texas, Southwestern, who was not involved in the study.
SYNGAP1 has other links to autism as well. For example, it regulates expression of the autism-linked gene MEF1. And it is a member of a family of genes that dampen activity of the RAS pathway, many of which, when mutated, also have links to autism.
To home in on the specific effects of SYNGAP1 mutations, the researchers engineered a series of mouse models that lack one copy of SYNGAP1 either in certain cells or at certain times in development.
Deleting one copy of SYNGAP1 in the entire mouse brain leads to social deficits, seizures and problems with learning and memory. Deleting the copy only in pyramidal neurons has the same effect, underscoring the importance of these neurons. By contrast, mice missing one copy of SYNGAP1 in inhibitory neurons of the forebrain look normal.
“This is a nice, simple demonstration that you don’t need to have deficits in inhibition to have excess of excitatory activity,” says Vikaas Sohal, assistant professor of psychiatry at the University of California, San Francisco. Being able to focusonly on excitatory circuits gives researchers a defined targetwhen it comes to treatments, he adds.
The researchers also engineered mice that express normal levels of SYNGAP1 in pyramidal neurons but are missing one copy in the rest of the brain. This alleviates the mice’s problems with memory and learning but not their seizures.
This finding is intriguing because it is often difficult to know whether cognitive deficits are a symptom in their own right or are the result of frequent seizures, says Huber. “As far as I know, that’s the first time someone has been able to dissociate a seizure phenotype from cognitive phenotypes,” she says. “This suggests there are different circuits that are mediating these two different types of behaviors.”
The study also found that enhanced signaling in pyramidal neurons tracks perfectly with memory problems in the mice. This suggests that researchers should correct this brain signal instead of watching for changes in behavior, which are harder to assess, says Rumbaugh. “Now the next phase of the project is to try and figure out how to make these animals better.”
1: Ozkan E.D. et al. Neuron 82, 1317-1333 (2014) PubMed
2: Clement J.P. et al. Cell 151, 709-723 (2012) PubMed