This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
Advances in research and help from families have brought scientists to the brink of an effective therapy for Angelman syndrome.

The autism community should be paying close attention to Angelman syndrome, a condition related to autism that is caused by mutations in a single gene.
Investments in basic and clinical research, along with the efforts of an intrepid community of families, has brought us to the brink of a ‘cure’ for the syndrome. If successful, these efforts could provide a blueprint for treating other conditions, including autism.
Angelman syndrome is characterized by a range of features, including developmental delay, motor problems, seizures, sleep problems and, in many cases, autism. Most people with the syndrome are minimally verbal, walk with a wide-spaced, unsteady gait (ataxia) and show repetitive behaviors such as hand-flapping. Despite these difficulties, they are frequently exuberant and happy.
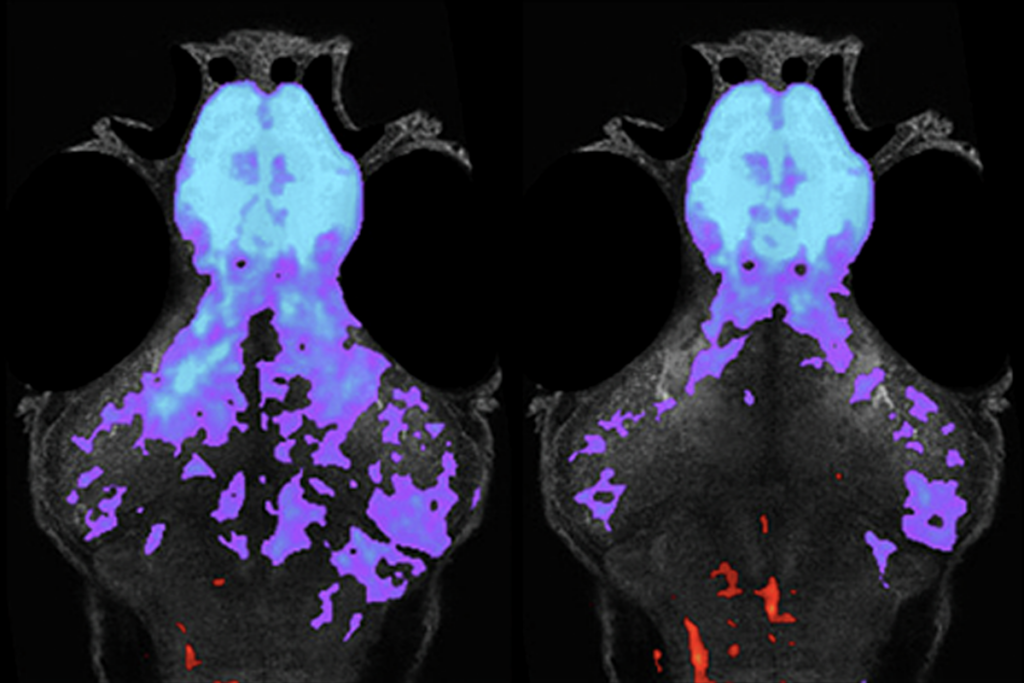
The gene mutated in the syndrome, UBE3A, holds the key to potential therapies. Only the copy of UBE3A inherited from the mother is typically active in neurons. In people with Angelman syndrome, this maternal copy is nonfunctional; the paternal copy is perfectly good — but silent.
Treatment strategies center on restoring expression of UBE3A by either replacing the maternal copy — gene therapy — or activating the silent paternal copy. Scientists have made progress on both fronts.
Directly replacing the gene may seem like the most straightforward approach. Viruses can deliver a gene as small as UBE3A to neurons efficiently. The challenge is to regulate how many copies of the gene are delivered to cells and how much protein is produced from each copy — because having too much of the protein can also cause problems.
Because of this, multiple academic groups and pharmaceutical companies are trying to carefully calibrate the gene dose as they develop gene therapies for Angelman syndrome. Among the teams at work on gene therapy for Angelman syndrome are those at the University of Texas Southwestern Medical Center, the University of Pennsylvania and PTC Therapeutics.
Another challenge is that, in people, the UBE3A gene can produce three slightly different proteins located in different parts of a neuron. One form is predominant, but it is not clear whether the other forms have specific roles. Gene therapy can likely replace only one form. So if the different forms of UBE3A have different functions, gene therapy may only be able to restore the function of one of the forms.
The second approach, activating the silent paternal copy of the gene, also seems feasible. In 2011, researchers established proof of principle of this approach. They screened about 2,400 small molecules to see if any of them could turn on the silent copy1.
One that did is a topoisomerase inhibitor, a blocker of enzymes that untangle DNA. Inhibiting these enzymes may halt the production of an RNA that silences UBE3A. However, topoisomerase inhibitors have side effects, because they influence other genes, too.
Small DNA-based drugs can do the same thing with more precision. These drugs are similar to nusinersen (marketed as Spinraza), a treatment for spinal muscular atrophy.
Nusinersen is an antisense oligonucleotide that boosts the expression of a vital motor neuron gene.
A few years ago, researchers tested an antisense oligonucleotide for Angelman syndrome. The molecule unsilenced paternal UBE3A and improved cognition in a mouse model2. Based on this work, three companies (Ionis, Roche and GeneTx) are pursuing similar therapies for the syndrome. These drugs are likely to require repeated spinal injections.
Another tactic involves CRISPR/CAS9, a programmable DNA editing system. Though the system works like topoisomerase inhibitors do, it is probably far more precise. Importantly, it can be packaged into a virus and delivered like other gene therapies. A range of RNA-cutting molecules can also be packaged into viruses to deliver the drug in a one-and-done fashion. Yet other methods include small molecules that activate paternal UBE3A by unknown mechanisms.
Clinical trials of some of these therapies could begin within a year or two. It is exciting to have multiple ‘shots on goal’ for Angelman syndrome.
Researchers and advocacy groups are discussing newborn screening and prenatal diagnosis so that everyone with Angelman syndrome has access to these therapies when they might be most effective. This Herculean effort would not be possible without the collaborative efforts of all corners of the community.
If we can develop a transformational therapeutic for Angelman syndrome — and it seems almost certain we will — it is likely to pave the way toward similar solutions for autism. To quote Booker T. Washington, “Success always leaves footprints.”
Stormy Chamberlain is associate professor of genetics and genome sciences at the University of Connecticut at Farmington.