Mice with mutations that affect the transport of a key cellular metabolite have altered neuronal and synapse structure and some autism-like traits, a pair of new studies show.
The metabolite, called acetyl coenzyme A (acetyl-CoA), modulates a quality-control pathway inside the endoplasmic reticulum — the cell’s protein-manufacturing hub — ensuring the destruction of any improperly folded proteins.
Mice with a mutation in SLC33A1, a gene that increases acetyl-CoA movement through this hub, have autism-like traits, the researchers showed in 2016.
Similar, though not identical, traits emerge in mice with mutations in two other genes — SLC13A5 and SLC25A1 — that, together with SLC33A1, ensure acetyl-CoA is transported properly to the endoplasmic reticulum, the team’s new studies show.
“These three mouse models prove that indeed a genetic connection in humans is real,” says Luigi Puglielli, professor of medicine at the Waisman Center at the University of Wisconsin-Madison, who led all three studies.
The genes and others like them may be associated with autism in people, some research has shown. For example, cells from people with the autism-linked condition 22q11.2 deletion syndrome also have altered SLC25A1 expression.
The endoplasmic reticulum interacts closely with energy-producing mitochondria to govern metabolism. The papers together shine light on the oft-ignored role of metabolism in brain development, says Victor Faundez, professor of cell biology at Emory University in Atlanta, Georgia, who led the 22q work but was not involved in the new studies.
“People are going to receive this with joy and, I think, skepticism,” Faundez says. “The skepticism is going to come from people that have dogmatic views about how you study the genetics of these disorders in animal models. I think this is a big contribution because it’s telling me that a process that has been neglected from study is affecting behavior in big ways.”
I
n the SLC25A1 work, published in Brain in February, the researchers overexpressed the gene in neurons in the forebrains of mice. The animals’ cells had increased levels of both acetyl-CoA and citrate — the driver of the cycle that produces cellular energy in mitochondria.The model mice showed behavioral differences from controls: They jumped repetitively in their enclosures and buried fewer marbles during a standard test of repetitive behaviors. In a memory test, they spent more time than controls investigating both novel and familiar objects, and they didn’t show the typical preference for a novel object. Their social interactions remained unchanged.
In a paper published in Brain Communications in January, mice with overexpressed SLC13A5 also showed the jumping behavior and buried few marbles. And unlike controls, they showed no preference for a novel mouse over a familiar one, showing an altered social behavior not seen in the SLC25A1 mice.
The differences between the models — and their deviations from many other autism model mice, which often show increased marble burying — are likely due to the fact that neither gene causes autism in people on its own, Faundez says. “It tells me it’s part of a package of genes that are modifying the behavioral output.”
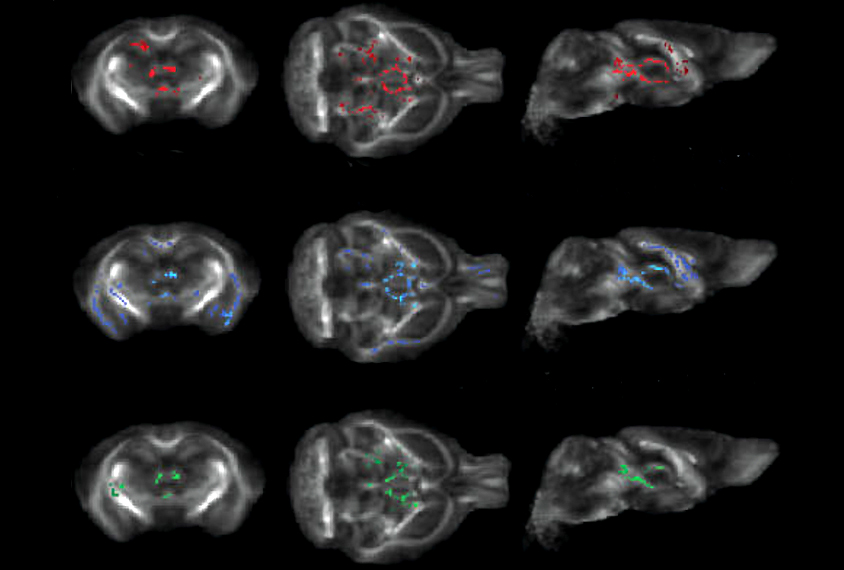
Compared with controls, the SLC25A1 mice had more microglia that showed a branching pattern associated with increased activation — which occurs in response to neuronal damage — even though the cells had typical levels of SLC25A1 expression. The model mice also showed structural changes in their white matter, but not in other cell types, including astrocytes and oligodendrocytes.
Neurons from the SLC25A1 mice appeared to mature more rapidly than those of controls and showed more branches, synaptic density and excitability.
The mutation altered expression of 99 proteins in the hippocampus and 227 in the cortex, as well as the proportion of 253 peptides tagged by acetyl-CoA in those brain areas. Many of the affected proteins and peptides are involved in processes associated with synapse development, the researchers found.
T
he papers support emerging literature that mitochondrial function is important for brain development and may be connected to autism, Faundez says.The findings fit with a theory that altered metabolism early in development can prevent neurons from maturing at the appropriate time, says Elizabeth Jonas, professor of internal medicine and neuroscience at Yale University, who was not involved in the work. Mitochondria from mouse models of fragile X syndrome fail to close a membrane leak that’s needed only in early development. And, similar to the SLC25A1 mice, fragile X mice show elevated citrate levels, Jonas has found.
The neurons in both studies are “growing too much; they’re growing too far; they’re making too many aberrant synaptic connections, because they’re still in that immature state, and that’s defined by this metabolism,” Jonas says. “We’re seeing this metabolism occur more and more in different settings.”
Puglielli says he next plans to further dissect the biochemical and metabolic reasons that the three model mice differ from one another, and to probe more genetic targets that affect the acetyl-CoA pathway.