This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
Loss of MeCP2, the Rett syndrome gene, in neurons that release the chemical messenger dopamine may lead to the motor deficits associated with the syndrome.
Loss of the Rett syndrome gene, MeCP2, in neurons that release dopamine — a neurotransmitter that regulates motor function and reward — may lead to the motor deficits associated with the syndrome, according to a study published 31 August in Neurobiology of Disease1.
Rett syndrome is an autism-related disorder that emerges around 6 to 18 months of age. Symptoms include social withdrawal, breathing difficulties and Parkinson’s-like motor deficits. Lacking one copy of the MeCP2 gene, which is located on the X chromosome, leads to Rett syndrome in females. This deletion is fatal in males, who have only one X chromosome.
In mouse models of the syndrome, male mice lacking the MeCP2 gene have more severe symptoms earlier in life and die at a younger age than do females with one copy of the gene. Researchers generally study the male mice because they have severe symptoms at a young age, but female mice better mimic the disorder in people, the study suggests.
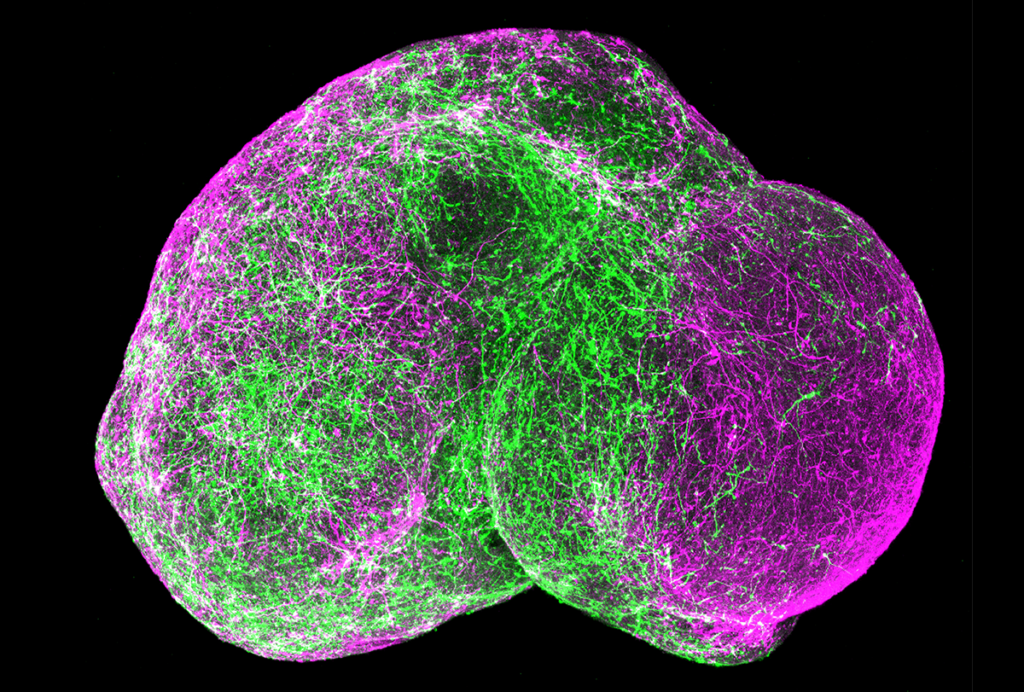
In the new study, the researchers looked at dopamine-producing neurons in the substantia nigra — a brain region that regulates motor function and is impaired in Parkinson’s disease — in female mice lacking one copy of MeCP2.
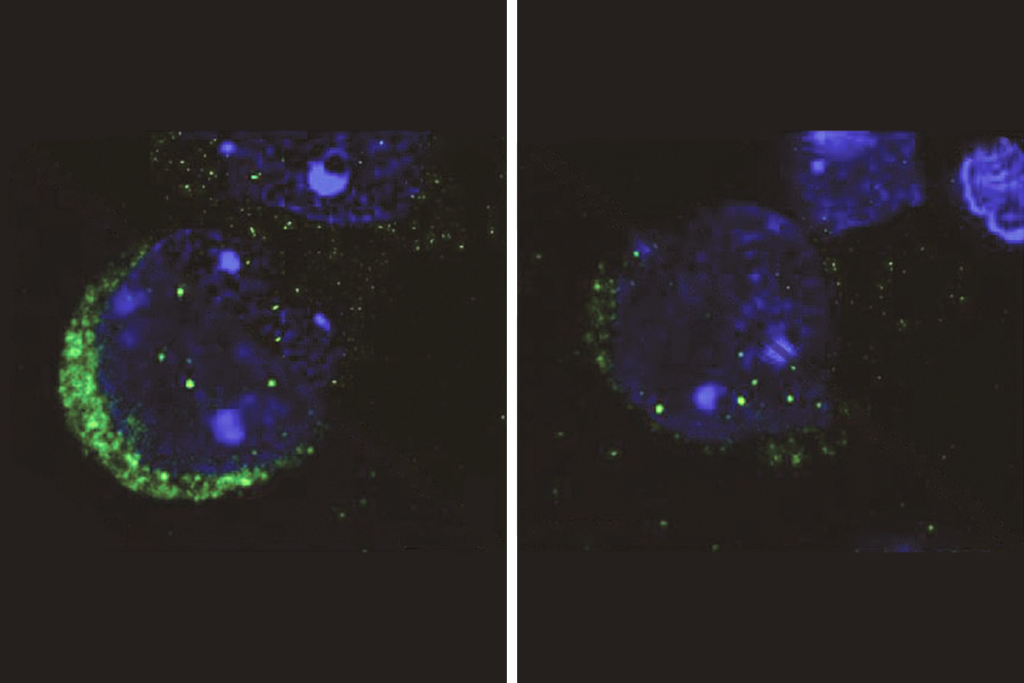
Because one copy of the X chromosome is randomly silenced in females in a process called X-chromosome inactivation, only a subset of dopamine neurons in these mice lack the MeCP2 protein, the study found.
These neurons are smaller and have fewer dendrites, the neuronal projections that receive signals from adjacent neurons, compared with other dopamine-releasing neurons in the same mice.
The MeCP2-deficient neurons also release less dopamine when stimulated than control neurons do. This effect is more pronounced in 1-year-old mice that show Parkinson’s-like symptoms than in 26-day-old mice that are asymptomatic.
This result adds to a 2010 study showing that MeCP2 mutations in a different subset of neurons — those that release gamma-aminobutyric acid, or GABA — are sufficient to reproduce nearly all the symptoms of Rett syndrome.
1: Gantz S.C. et al. J. Neurosci. 31, 12629-12637 (2011) PubMed