Huntington’s disease gene variants past a certain size poison select cells
The findings—providing “the next step in the whole pathway”—help explain the disease’s late onset and offer hope that it has an extended therapeutic window.
More than 150 years after the first known description of Huntington’s disease and 32 years after the causative gene, HTT, was identified, new evidence has emerged to explain how variants linked to the disease devastate the brain: The toxicity comes not from the initial variant itself but rather from its dynamic expansion past a set threshold in specific cells, according to a study published today in Cell.
The results help explain why most people with Huntington’s disease don’t start to show symptoms—including muscle rigidity, irregular movements and severe psychological issues—until age 30 to 50, with the gradual loss of striatal projection neurons, also called medium spiny neurons, says co-lead researcher Steven McCarroll, professor of biomedical science and genetics at Harvard Medical School. “We hadn’t been thinking about mutations as dynamic things” that become toxic only later in life, he says.
The HTT variants associated with Huntington’s disease all have extra repeats of the DNA triplet CAG. Typical people carry about 15 to 30 of these repeats, and those with the disease tend to have 40 or more. The disease-linked expansions, which are known to grow even larger over time, result in a gangly version of the Huntington’s protein that is thought to cause neurons to malfunction and degenerate.
But the expansion does not appear to affect a cell’s biology until it exceeds 150 CAG copies, according to the new study. And the repeats accumulate quietly over the course of years, and at different rates for different cells.
Striatal projection neurons with more than 150 repeats have severely dysregulated transcriptomes, McCarroll and his colleagues found by analyzing gene expression in postmortem tissue from people with Huntington’s disease. But other cell types in the striatum, including oligodendrocytes and interneurons, do not end up with as many repeats, nor do they undergo similar transcriptomic changes, the work shows.
“It’s one of the most important studies that’s come out since the genome-wide association studies,” which suggested that the disease is driven by expansion of the CAG repeat, says Gillian Bates, professor of molecular neuroscience at University College London, who was not involved in the new work. “It’s the next step in the whole pathway.”
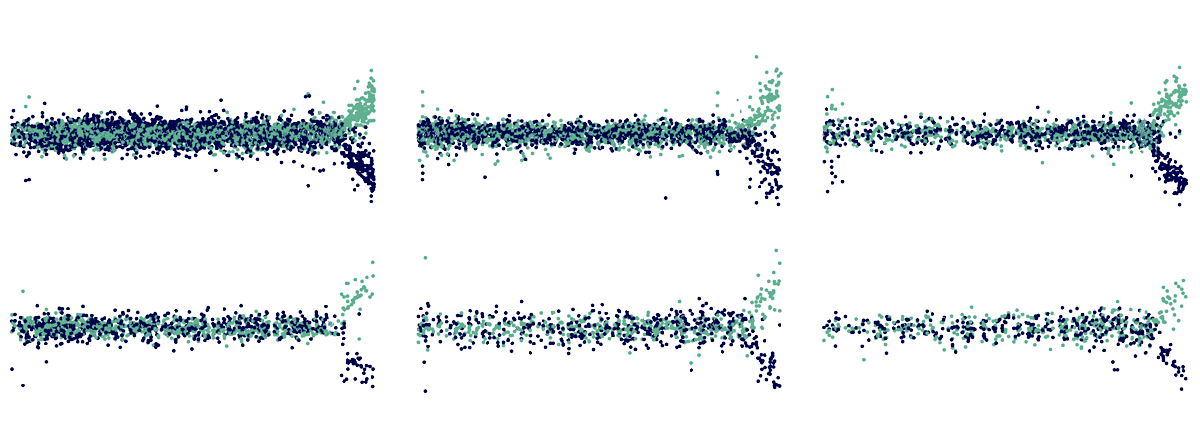
Danger zone: Beyond 150 CAG repeats, multiple genes within striatal projection neurons, shown here from six different donors, become up- or down-regulated (teal, blue, respectively).
Linking the repeat lengths to an individual cell’s transcriptomics is an important finding, agrees Christopher Pearson, senior scientist in genetics and genome biology at the SickKids Research Institute, who was not involved in the work. But whether repeats of fewer than 150 copies are truly harmless remains controversial, he says.
“They aren’t looking at function—whether this is a happy cell or a sick cell,” Pearson says. Before reaching 150 repeats, the transcriptomics may not be pointing to cell death, he adds, “but that doesn’t mean that those cells aren’t sick.”
T
he CAG repeats in disease-causing HTT alleles are prone to somatic expansion with age: New copies are likely introduced when, during transcription, the two separate strands of DNA become incorrectly paired as they reunite. In fact, additional variants in DNA-mismatch-repair genes correlate with an especially early or late onset of Huntington’s disease, according to a 2019 genome-wide association study—suggesting that an increase or decrease in mismatches, and thus repeats, influences the pathology.
Following that lead, McCarroll and his colleagues developed a new approach to measure a cell’s repeat length alongside its RNA expression. Striatal projection neurons in the brain of someone with Huntington’s disease can accumulate more than 800 repeats, they found. And the number of repeats varies greatly from cell to cell, lining up with results from a 2003 study on human striatal and cortical cells.
Striatal projection neurons with more than 150 repeats have genes that are atypically up- or down-regulated, which results in a loss of the cells’ identity, single-nucleus RNA sequencing in the new study revealed. In cells with the most dysregulated transcriptome, genes such as CDKN2A and CDKN2B, which are involved in cell death, are highly expressed.
The repeat expansion is “a slowly ticking DNA clock,” McCarroll says. Individual cells tend to gradually gain “biologically innocuous” CAG repeats for decades with no effect to their transcriptomes. But once the expansion surpasses the threshold of 150 repeats, the decline happens rapidly for that cell, a model of the progression of repeats over time showed. The model used data the team collected from the postmortem brains of 50 people with and 53 people without Huntington’s disease.
T
he sharp change McCarroll and his colleagues found in a cell’s transcriptome beyond 150 repeats contradicts other recent findings. Cells in human and mouse tissue that have fewer than 150 CAG repeats still show dysregulated gene expression, previous studies suggest. And other cell types also exhibit somatic expansion and atypical transcriptomes in Huntington’s disease but remain resilient to cell death, according to a 2024 study.
But these other studies focused on CAG repeats only up to 150, because of technological limitations in sequencing highly repetitive stretches of DNA. The new work implemented a different, more time-consuming method that enabled the team to measure the longer repeats. That difference in approach may partially explain the discrepancy between the new and previous findings, Pearson says: Other studies were not looking at the gene expression in the most extreme cases.
Still, it is not clear why the new study saw no pathological effect from fewer than 150 repeats, Pearson says.
The new study also does not investigate what it is about 150-plus CAG repeats that disrupts gene expression or why striatal projection neurons are particularly vulnerable, Bates says. But it does have exciting implications for the disease’s treatment, she adds: If a therapeutic approach can halt or slow a cell’s somatic expansion, perhaps it can prevent the cell from ever becoming pathogenic. Even once symptoms begin, “most of the cells won’t have entered into that phase,” potentially leaving a fairly large therapeutic window, she says. “It’s a big deal.”
Multiple companies are working on targeting mismatch repair pathways to slow the expansion process. And such treatments may be relevant for other conditions caused by DNA repeat expansions, such as spinocerebellar ataxia, Pearson says.
The work validates the importance of paying attention to common variants in human genetics results, even when they seem to show only a small effect, McCarroll says. The 2019 genome-wide association study pointed to changes in mismatch repair genes—some of which postponed the onset of Huntington’s symptoms by only a few months, he says. “And yet we were sure that they were trying to tell us something.”