Although the brain has been an object of fascination for centuries, neurobiology as we know it originated with Santiago Ramon y Cajal’s magnificent descriptions and drawings of neurons in the late 1800s. His work, which spanned nearly every part of the nervous system of dozens of invertebrate and vertebrate species, set the field’s agenda for the next hundred years: the detailed analysis of single neurons, initially morphologically and later electrophysiologically. It was the first single-cell revolution.
By the end of the 20th century, adherence to this single-cell agenda — coupled with advances in molecular methods, including biochemistry, molecular biology, immunochemistry and transgenesis — had revealed the basics of neuronal structure, function and development. Despite these transformative advances, however, it became increasingly clear that to truly understand how the brain works — and how it fails in neurological and psychiatric diseases — would require new approaches. To make sense of circuit architecture and pinpoint cells that might be defective in brain diseases, for example, researchers would need to be able to study enough single neurons to classify them into types. That would require the capacity to analyze hundreds to thousands of single cells, preferably simultaneously.
That has finally become possible, with the invention over the past 20 years of a series of massively parallel high-throughput single-cell methods. Today, researchers can molecularly profile cells using high-throughput single-cell and single-nucleus RNA sequencing (scRNA-seq and snRNA-seq). They can monitor neuronal activity with multi-electrode “neuropixel” type probes and genetically encoded calcium and voltage indicators. They can visualize neuronal structure and connectivity with serial-section electron microscopy or optically with super-resolution and expansion microscopy. In each case, they can assay hundreds to thousands of cells in parallel sufficiently quickly and inexpensively (relatively speaking) to classify and characterize neurons and unravel neural circuitry more comprehensively than ever before.
In other words, we are now in the midst of a second single-cell revolution.
This new capacity to accurately classify cell types on a broad scale is revolutionizing how we study neural circuits, providing new insights into brain development and evolution, and opening new avenues for understanding brain diseases. But to fully realize its potential, the field still needs to grapple with a number of questions, such as what the most appropriate level to classify cells is and how closely different facets of cell-type data align. This introductory essay sets the stage for an ongoing series that will examine the uses of this technology for neurobiology, along with challenges that remain.
O
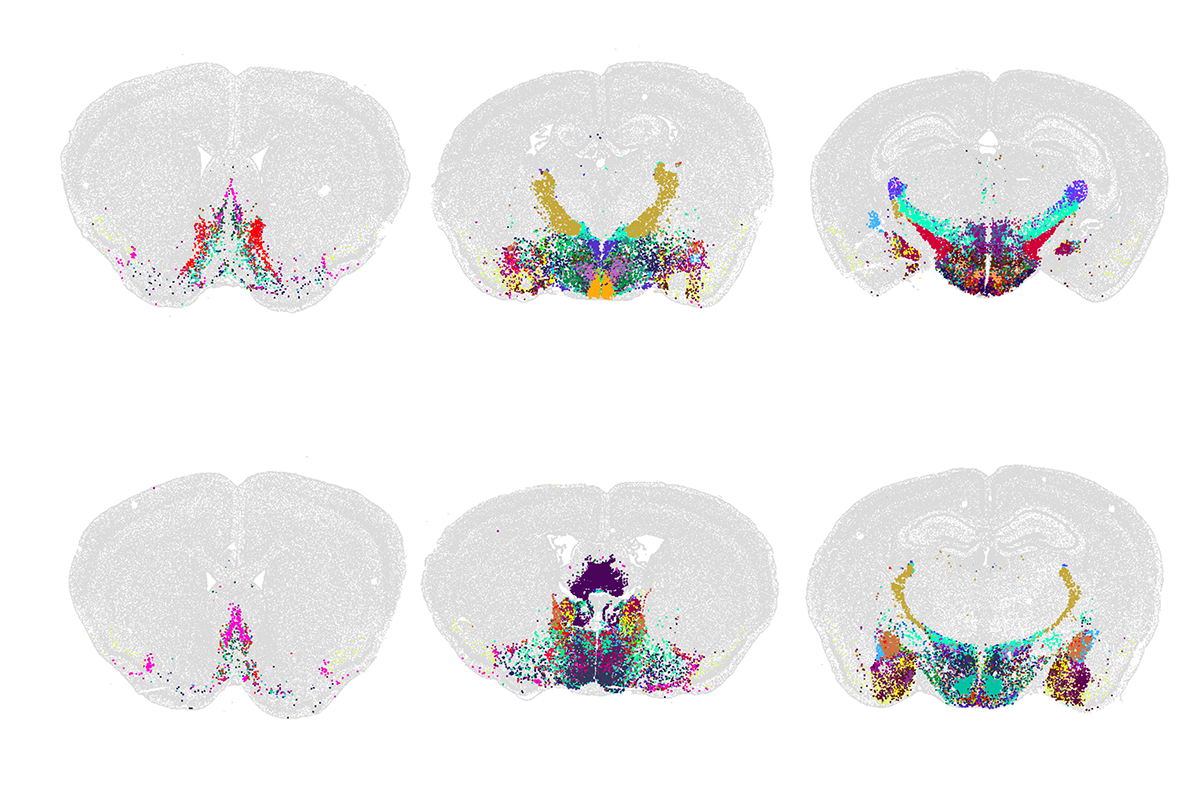
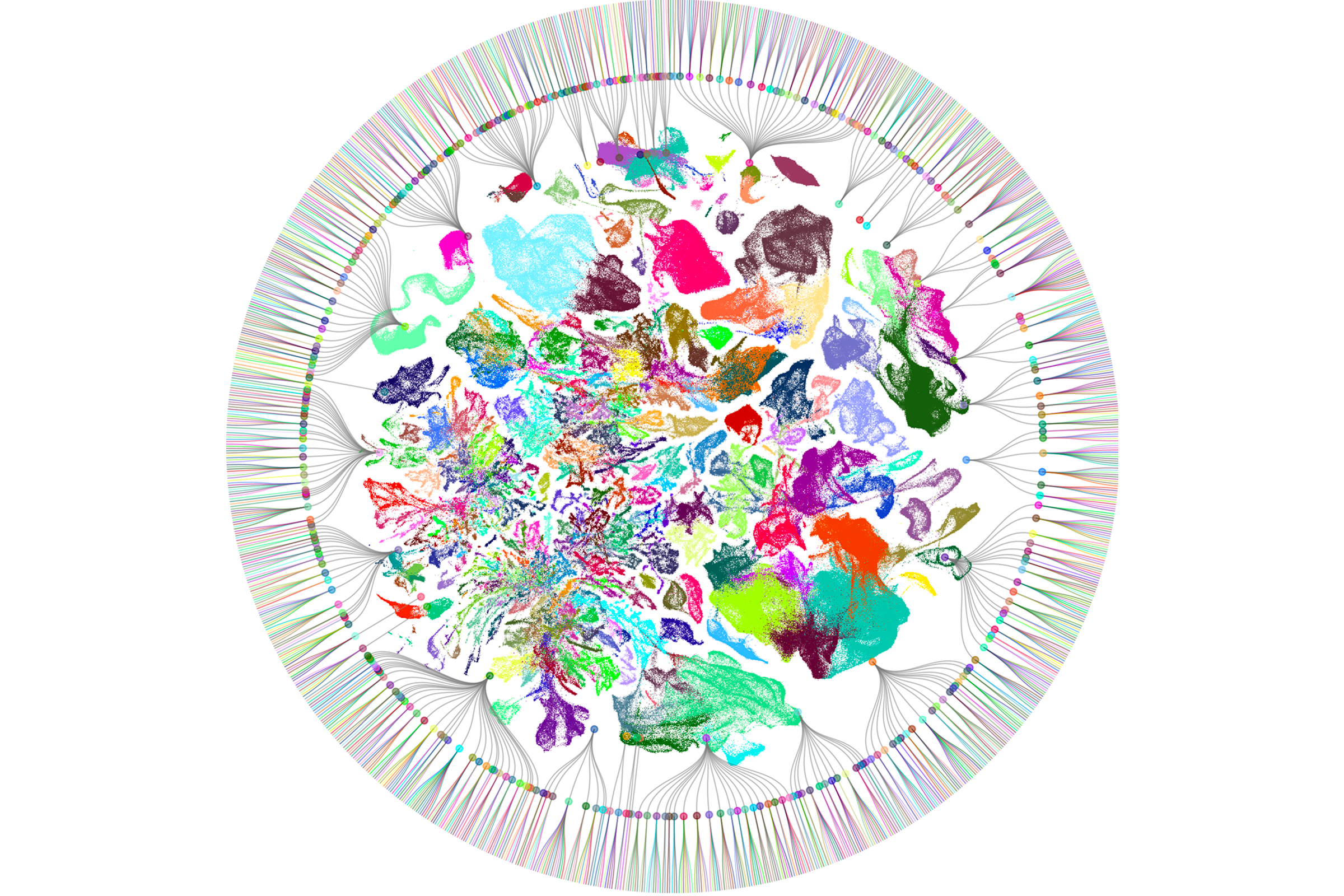
f these new high-throughput methods, none has had a greater impact than sc/snRNA-seq. The method was invented independently and nearly simultaneously by three groups almost 10 years ago — Drop-seq by Evan Macosko and his colleagues, inDrop by Allon Klein and his colleagues, and 10X/GemCode by Grace Zheng and her colleagues. In all three platforms, mRNA from thousands of single cells or nuclei is captured and barcoded, then reverse-transcribed, amplified and sequenced in a single reaction. Cells with similar transcriptomes can be computationally grouped into candidate cell types. Newer methods provide similar results at an even lower cost than the original techniques. By now, researchers have profiled more than a billion single cells.The first successful effort to use scRNA-seq to generate cell-type atlases of complex tissues used the mouse retina, a particularly accessible part of the brain; we now know that it contains some 130 neuronal types. Researchers have since applied the method to numerous other tissues and species. Over the past few months, Science and Nature have published special issues detailing the largest results from these efforts to date, including expansive atlases of the human and mouse brain. (For more, see “Vast diversity of human brain cell types revealed in trove of new datasets.”)